MOLECULAR RESULTS
PHKA2 Gene sequencing: POSITIVE for a hemizygous deletion in exon 33 of c.3648_3649delAA, which results in a frameshift and the development of a stop codon 26 codons downstream (p.Arg1217SerfsStop26).
AGL Gene Sequencing: Heterozygous Variant of Unknown Significance of c.1481 G>A. This causes a single amino acid substitution (p.Arg494His).
ALDOB sequence and deletion/duplication analysis: NEGATIVE.
FBP1 Sequence Analysis: NEGATIVE.
DISCUSSION
Glycogen storage diseases result from the inability to properly metabolize glycogen. Glycogen is a carbohydrate which is able to be stored and easily mobilized to maintain appropriate blood sugar levels during periods of fasting. This molecule is most abundant in the liver and skeletal muscle; this localization contributes to many of the observed clinical symptoms. Symptoms of glycogen storage diseases include muscle pain, cramps, exercise intolerance, and easy fatigability. Signs of fasting hypoglycemia, ketosis, and poor weight gain, with or without hepatomegaly, can also be seen1. Glycogen storage diseases have a prevalence of approximately 1/10,000, although their frequency is believed to be underestimated due to the lack of efficient molecular methods2.
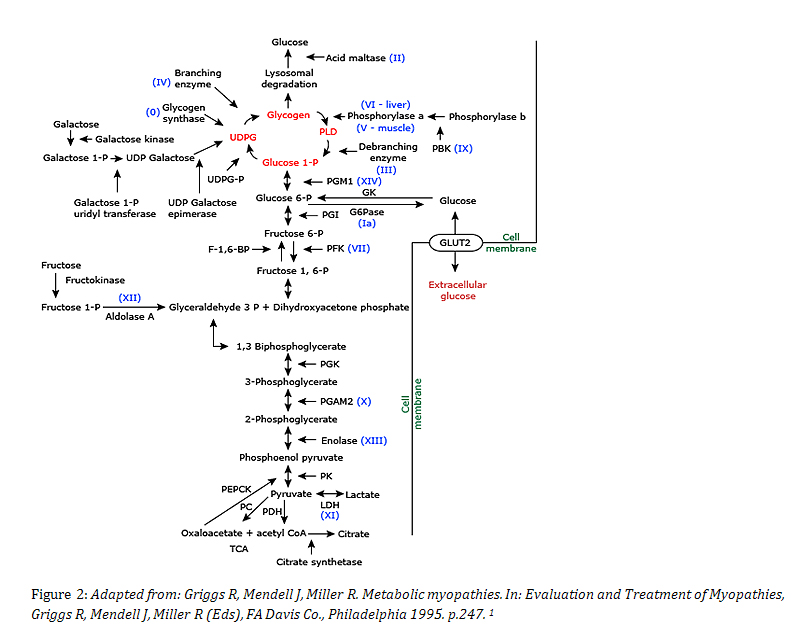
There are numerous pathways involved in glycogen metabolism, and defects in any of them can cause different types of glycogen storage disease, as demonstrated in this graphic (Figure 2):
The mutation seen in the presented case involves a two nucleotide deletion in the PHKA2 gene. This gene encodes for the alpha subunit of the liver isozyme of phosphorylase kinase B3. The phosphorylase kinase (PHK) enzyme is important in the breakdown of glycogen to Glucose-1-P and dysfunction can result in GSD Type IX. PHKA2 is encoded on the X-chromosome, and as such GSD-IX is also referred to as X-linked glycogenosis (XLD)4, 5. The PHKA2 enzyme consists of 4 subunits, and the alpha subunit is a 138kDa protein that has an inhibitory role within the PHK complex6, 7. This gene is usually the first sequenced when there is concern for GSD-IX, as it is dysfunctional in 75% of cases4.
Patients with type IX GSD often have excessive glycogen in the liver and can present with short stature, hepatomegaly, and sometimes ketotic hypoglycemia4. They often show signs of transaminase and triglyceride elevation, but cirrhosis is very rare4. Enzymatic studies assaying for the activity of the PHKA enzyme can be performed in vitro, however these often show negative results even when the in vivo enzyme is dysfunctional4, 6 ,8. Molecular studies are ordered to rule out this disease and to prevent more invasive studies (such as liver biopsy) 6, 7, 8, 9. This case demonstrates a deletion of two adenine nucleotides at position 3648-3649 of exon 33 in the PHKA2 gene, which results in an amino acid change and subsequent frameshift. In this patient, the frameshift actually results in a longer protein, with a carboxy terminal sequence change as follows (the altered amino acid sequence is in bold):

This amino acid sequence is obviously significantly altered from the above wild type, and the function is likely to be impacted by these changes.
Most frameshifts result in the formation of an early stop codon, and subsequent truncation of the protein. Truncated proteins tend to get degraded within the cell, and thus result in loss of enzymatic activity in vivo6, 7. This specific microdeletion (and subsequent frameshift) has been reported in a patient from Poland, and resulted in mild fasting intolerance in addition to hepatomegaly10. As most PHKA2 gene mutations are found in specific families, with few uniformly present in populations, it is relatively surprising that this microdeletion has been previously reported8. Also of note, as demonstrated by the pedigree on the prior screen, there have been very few males born in this family. As this mutation is X-linked, and since there are no de novo mutations reported in the literature, it seems likely that this mutation has been carried in the females in this family over a long period of time before revealing itself in this patient.
The second variant identified in this patient, c.1481G>A of the AGL gene, results in an Arginine to Histidine amino acid change at position 494. The AGL gene encodes for the glycogen debranching enzyme (GDE), which is required for glycogen breakdown and subsequent phosphorylation11, 12. Partial or total lack of this enzyme results in GSD Type III, which is autosomal recessive, and felt to be rare11. In patients lacking this enzyme, glycogen accumulates in the liver, heart, and muscle11. Clinical manifestations include hepatomegaly, hypoglycemia, short stature, and sometimes myopathy and cardiomyopathy12. Mutations in AGL are variable, and many are confined to single families9.
This protein change is predicted to be "probably damaging" by the PolyPhen-2 protein prediction software, with the SIFT software program predicting this variant as "not tolerated." This variant has also been previously identified, but only in the context of a patient with two other known damaging mutations13. Thus, the clinical significance of this variant is unclear. Given the patient's more mild clinical presentation, it is likely that this variant is not significantly contributing to his disease.
This case demonstrates the difficulty in interpreting multiple genetic variants in the same patient, especially when these genetic changes involve similar pathways and present with similar symptoms. Performing reflex testing to ensure that only the appropriate testing is performed, in an order agreed upon by the clinical team, may help alleviate some of these redundant and potentially confusing results. However, given the future of personalized medicine, multiple variants of unclear significance may be revealed in patients, even if other pathogenic mutations have already been identified. Thus, the interpretation of these results will likely continue to prove difficult, with further genomic study hopefully helping to identify rare benign SNPs as opposed to truly pathogenic alleles.
REFERENCES
![]() Contributed by Ryan A. Collins, MD and Marie DeFrances, MD, PhD
Contributed by Ryan A. Collins, MD and Marie DeFrances, MD, PhD