FINAL DIAGNOSIS
Positive for Huntington Disease at the high end of the reduced penetrance range.
Clinical Presentation of Huntington Disease
Huntington Disease (HD) is an autosomal dominant disorder which affects between 5 to 10 people per 100,000 population. It is most frequent in Europe and North America, but familial cases are documented worldwide [3]. HD symptoms usually appear during middle age (around 40 years of age) but juvenile onset cases (<20 years old) account for 5-10% of the cases. Late onset (>50 years of age) HD accounts for ~20% of cases. Disease progression generally occurs over the course of 15 to 20 years [4]. The major manifestations of HD are psychiatric, motor and cognitive dysfunction. Psychiatric symptoms include depression, anxiety, apathy and irritability. This may occur up to 20 years before any motor deficits are noticed; however, psychiatric symptoms may not be recognized as part of the disease process until later in the course. Motor dysfunction onset is usually marked by involuntary movements of the face, fingers, feet or thorax. Progression continues to choreiform movements of the head, neck, arms and legs. Cognitive impairments of memory and language comprehension worsen with disease progression. Weight loss is also present later in the disease and may be due to a combination of dysphagia and the degeneration of hypothalamic neurons which contain orexin to stimulate hunger. Towards the end of the disease course, patients become rigid and akinetic with severe dementia and mutism [3].
Huntingtin Protein
HD is caused by the expansion of the polymorphic CAG trinucleotide repeat in exon 1 of the HTT gene which encodes the huntingtin protein on 4p16.3. The HTT gene was discovered by the Huntington's Disease Collaborative Research Group in 1993. The discovery of the gene allowed for the more accurate predictive testing using PCR sizing. Huntingtin is a widely expressed protein with the highest levels in the brain and testis. It is found predominately in the cell cytoplasm but is also seen in the nucleus. Huntingtin interacts with many proteins that are necessary for appropriate gene expression, intracellular transport, intracellular signaling and metabolism. It also appears to have some anti-apoptotic properties demonstrated by a study showing neurodegeneration following inactivation of the gene in adult mice [3].
Prediction of the age of onset for a specific CAG repeat size
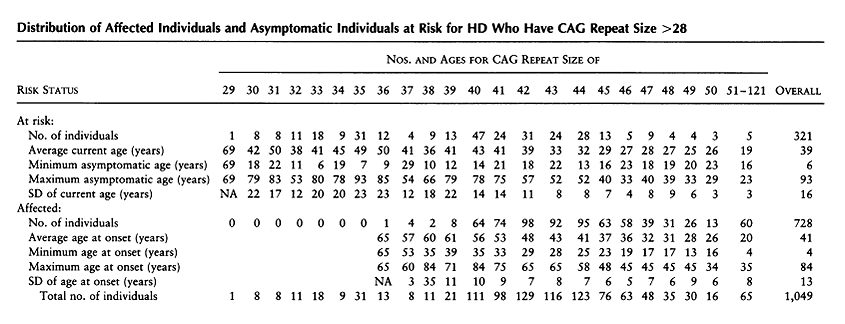
A major area of research following the discovery of the HTT gene centered around the idea that the size of the CAG repeat correlated with the severity of the disease and age of onset. Numerous studies showed that there was an inverse relationship between age at onset and the number of CAG repeats. However, no studies had included individuals with expanded alleles who were still asymptomatic. Brinkman et al conducted a study in 1997 which looked at 321 at-risk individuals and 728 affected individuals with an expansion size of 29-121 repeats. They noted complete penetrance for expansion sizes ≥42 repeats. They also found that only a proportion of those with CAG repeats of 36-41 would manifest symptoms within a normal life span. For patients with a CAG repeat of 39, 13 patients were asymptomatic, ranging in age from 12 to 79 years old. 8 affected patients were included, with an average age of onset of 61 years (see Table 2). Survival curves were significantly different for each CAG repeat size, supporting the hypothesis that expansion size is a dominant factor in determining the age of onset. Table 3 illustrates the probability of onset at different ages for a given CAG repeat size. Investigators also found that the difference in a single CAG repeat length has a significant effect on the expected age at onset for an individual with the median age at onset decreasing by 3.4 years for each CAG repeat increase in the 39-50 year range (Table 4) [4].
Table 2: Brinkman et al, 1997
Table 3: Brinkman et al, 1997
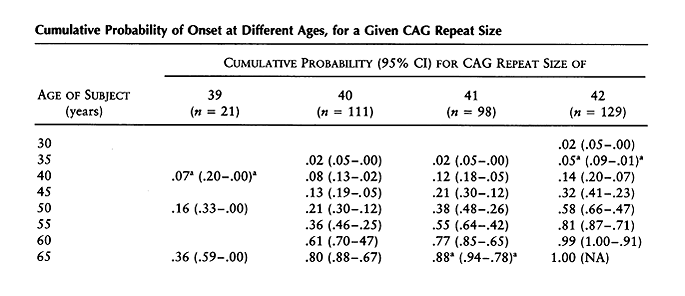
Table 4: Brinkman et al, 1997
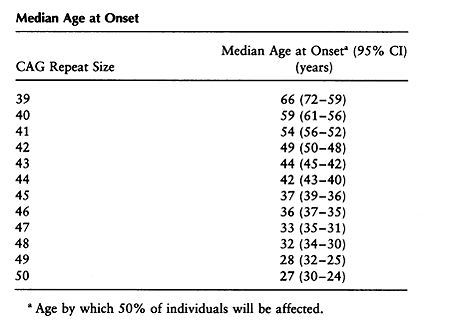
Table 5: Conditional Probability of Age of Onset in an Individual with 39 repeats. Langbehn et al, 2004, http://www.cmmt.ubc.ca/clinical/hayden.
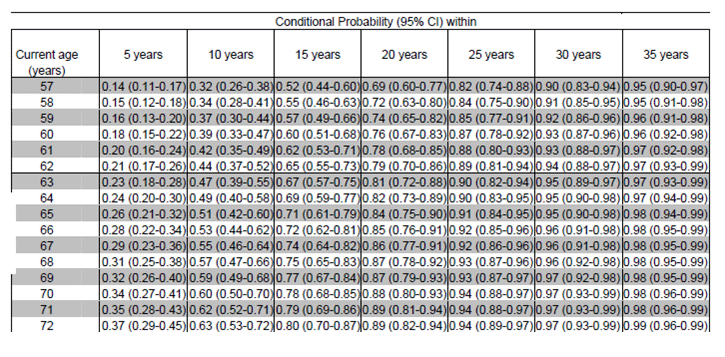
A new model, based on the original data of Brinkman et al, was published in 2004 and included a larger sample than the previous study but used the same methods for determining the length of the CAG repeat and clinical status. Information was available from 2913 individuals with CAG repeat lengths between 41 and 56 and was used to extrapolate information about smaller lengths. This study confirmed reduced penetrance in individuals with fewer than 40 repeats and showed that many individuals with <41 repeats will not show symptoms, including up to 86% of those with 36 repeats. Age at onset demonstrated greater variability with smaller CAG lengths than with higher repeat lengths. Thus, most individuals with larger numbers of repeats will experience disease onset within a narrow age range. Using a parametric model, these investigators composed a booklet of conditional predictions with mean and median ages of onset for individuals with CAG repeats between 36 and 56 in number. Table 5 is the conditional probability of onset for an individual with 39 repeats, such as our patient. Other genetic and environmental modifiers were hypothesized to be less significant in those with greater than 44 repeats and more important in smaller CAG repeat lengths. Additional predictors of age at onset in this study were paternal age of onset and maternal versus paternal transmission. Other research examining the interaction of the normal allele with the expanded allele has shown some interaction and other genetic polymorphisms may play a role [5].
Intergenerational expansions
The observation that paternal descent played a major role in juvenile HD led to several studies focusing on intergenerational expansions. Kremer et al. evaluated 254 affected parents and offspring with expanded CAG ranges. They found that CAG lengths were often unstable; contraction was observed in 18% of cases and expansion in 52% of cases. The majority (90%) of transmissions spanned a loss of 4 repeats to a gain of 7 repeats. CAG repeats increased in size when transmitted paternally in 68% versus 39% maternally. Moreover, large expansions (>7 CAG repeats) were predominately transmitted paternally. Mothers were more likely to transmit a stable or contracted repeat allele. A high correlation between repeat instability and paternal transmission was noted, but no such correlation was seen in maternal transmission. This was also influenced by a small number of large expansions transmitted from fathers with a large number of repeats. This study supported the observations that affected fathers are more likely to have offspring with large expansions than affected mothers. It also demonstrated that larger CAG repeats are more likely to have expansions during paternal transmission [6].
Psychological Aspects of Predictive Testing
With improvements in predictive testing methods for HD, the psychological consequences of this testing has gained major interest. Not only are the current studies not able to predict disease onset accurately in the lower range of expanded repeats, it appears that other genetic and environmental factors are involved. In a worldwide questionnaire study, 0.97% of participants had a catastrophic event following predictive testing. Of those individuals with a catastrophic event, 55% were symptomatic at the time of the event. All who committed suicide were symptomatic. The onset of disease has been previously recognized as a high-risk time for suicide attempts. The majority of patients with catastrophic events were given an increased risk result; however, 0.3% of patients with a decreased risk or no expansion also had catastrophic events following predictive testing. The mean age of predictive testing was age 37, indicating that reproductive decisions had already been made. The majority of individuals participating in predictive testing were female [7].
What does this mean for our patient?
Our patient has one normal allele and one expanded allele, with the # of CAG repeats calculated at 39 and 40 in replicates tested. A relevant question is whether the full penetrance category begins at ?40 repeats when it has been documented that occasional patients with 40 or 41 repeats will not manifest symptoms. Another issue arises from rounding differences for the # of CAG repeats between replicates. In this case, data looked somewhat more robust for the result of the 39.3 repeats, rounded down to 39 and that value was reported. The other replicate tested as 39.6 (rounded to 40) repeats. Should the reduced or full penetrance range be used in counseling this patient? An additional factor to keep in mind is the response of patients to negative (instead of positive) testing results. It is well known that some individuals feel significant guilt after testing negative for HD, particularly when other family members test positive. There is a report of a patient who received negative results with predictive testing developed depression and significant ballistic movements and was then diagnosed with HD. Direct-testing was performed confirming the normal CAG length, but this was followed by a suicide attempt [7]. The psychological aspects of predictive testing are complex. Our patient is already experiencing what she feels is a significant change in her ability to walk. The patient is also reported to have 6 children, although the medical record does not state their ages or whether or not they have children. It will be of interest to see how many of the relatives will proceed with predictive testing.
REFERENCES
![]() Contributed by Anna Woodard, MD and Jeffrey A. Kant, MD, PhD
Contributed by Anna Woodard, MD and Jeffrey A. Kant, MD, PhD