Final Diagnosis -- Fundic gland polyps with low-grade dysplasia
FINAL DIAGNOSIS: FUNDIC GLAND POLYPS WITH FOCAL LOW-GRADE DYSPLASIA.
DISCUSSION
Fundic gland polyps (FGPs) are the most common gastric polyps in the general population, as well as in patients with Familial Adenomatous Polyposis (FAP) 1,2. FGPs present usually as multiple and small polypoid nodules in the gastric fundus and body. Microscopically they are characterized by microcysts lined by fundic epithelium including oxyntic cells, and are usually lined by a shortened foveolar epithelium. The incidence of FGPs is 1.9% in the general population, 84% in FAP patients and 93% in Attenuated Familial Adenomatous Polyposis (AFAP) patients 2.
|
As a reminder, FAP is a disease caused by germline mutations in the APC (Adenomatous polyposis coli) gene, developing hundreds of adenomatous polyps in the colon. AFAP is a disease caused by germline mutations in the 5' or 3' end of the APC gene, and is also different clinically as it presents 10-15 years later than FAP and is associated with smaller number of colonic adenomas2.
|
Initially, FGPs were described as hamartomatous benign lesions with no potential for malignant transformation. However, recent literature demonstrate that they may harbor dysplastic foci or even malignant transformation 1, 2.
FGPs have different genetic alterations whether they appear in a patient with previous familial adenomatous polyposis (syndromic FGPs) or whether they arise sporadically 1.
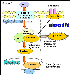 Syndromic FGPs arise through somatic mutations in the second allele of the adenomatous polyposis coli (APC) gene, whereas sporadic FGPs are caused by beta-catenin gene mutations. Although they are two different genes (APC and beta-catenin), they act through a common pathway, the Wnt-APC-beta-catenin pathway (Figure 5). Genetic mutations in these genes lead to deregulation in the activation of the Wnt-APC-beta-catenin pathway, which is responsible for both the syndromic and the sporadic FGPs.
Syndromic FGPs arise through somatic mutations in the second allele of the adenomatous polyposis coli (APC) gene, whereas sporadic FGPs are caused by beta-catenin gene mutations. Although they are two different genes (APC and beta-catenin), they act through a common pathway, the Wnt-APC-beta-catenin pathway (Figure 5). Genetic mutations in these genes lead to deregulation in the activation of the Wnt-APC-beta-catenin pathway, which is responsible for both the syndromic and the sporadic FGPs.
|
Alterations in the WNT pathway have a role in colorectal cancer. Inactivation of the second APC allele occurs in patients with FAP, who develop thousands of polyps in the colon that can progress to carcinoma. Somatic mutations in the APC gene have been also detected in 80% of sporadic colorectal polyps and carcinomas. Mutations in the beta-catenin gene have been found in 50% of colorectal cancers without APC mutations, and predominantly occur in association with a replication error phenotype (microsatellite unstable tumors).
|
Wnt-APC-beta-catenin pathway (Figure 5): Wnt genes encode for the Wnt glycoprotein, which regulates cell growth, motility and differentiation of cells during embryonic development. When the Wnt protein reaches a cell, it binds to transmembrane proteins located in the plasma membrane. These transmembrane proteins (named frizzled receptors) are then activated and signals are release to the intracellular space. Here the signal is transduced, leading to accumulation of stabilized cytosolic beta-catenin. Stabilized beta-catenin is translocated to the nucleus where it binds transcription factors of the T-cell factor family (TCF/lymphocyte enhancer factor family).
In the absence of Wnt protein, the beta-catenin is attached to the plasma membrane where it associates with E-cadherin molecules. The beta-catenin that does not attach to E-cadherin is trapped by a multiprotein-complex consisting of APC (Adenomatous polyposis coli) and other proteins. This multiprotein-complex degrades beta-catenin with the help of the proteasome. When APC mutations occur, the multiprotein-complex does not work and this leads to accumulation of beta-catenin in the cytosol 6, and increased available beta-catenin for nuclear transcriptional activation, leading to altered cell-proliferation, migration, differentiation and apoptosis, which contribute to tumor development 1,6.
Dysplasia in FGPs
Dysplasia can develop in FGPs and is more commonly found in the syndromic than in the sporadic forms. FAP-associated FGPs frequently show epithelial dysplasia, and neoplastic progression of FAP-associated FGPs in the form of large dysplastic polyps or invasive adenocarcinoma has been reported. In contrast, sporadic FGPs with their predominance of beta-catenin mutations, only infrequently show dysplasia and have not been reported to progress to adenocarcinoma.
Dysplasia occurring in FGPs is associated with mutations in the APC gene 2, whereas beta-catenin mutations do not frequently lead to dysplasia in FGPs. A recent study has demonstrated that dysplasia in sporadic FGPs is more likely to be associated with APC gene mutations than with beta-catenin mutations. This means that sporadic FGPs with dysplasia are molecularly more similar to FAP-associated FGPs 5, and that a clonal genetic alteration is involved in the development of FGPs. Thus, FGPs have the potential to become neoplastic lesions.
|
As in our case, low-grade dysplasia has been observed in up to 53% of the syndromic FGPs, and in up to 2.3% of the sporadic FGPs 2, 4. In other series, including a study of 319 fundic gland polyps, the FAP-associated polyps showed low-grade dysplasia in 25%, and the sporadic ones in 1% of the cases 3, 8.
|
Low-grade dysplasia criteria, include nuclear enlargement, stratification and hyperchromatism in the foveolar or surface epithelia overlying FGPs. The reason why the FGPs of FAP patients have a higher prevalence of foveolar and surface epithelial dysplasia is not fully understood. In these cases, the epithelium of the subjacent dilated fundic glands is not dysplastic, and no epithelial dysplasia, intestinal metaplasia or glandular atrophy is identified in the non-FGP fundic mucosa. The clinical significance of foveolar and surface epithelial dysplasia in FGPs is unclear. Regression of dysplasia in these lesions has been previously reported in 36% with mild gastric dysplasia, although 21% progressed to severe dysplasia 3, 9.
FGPs with no dysplasia do not differ in beta-catenin, p53 or Ki-67 expression, as compared to fundic mucosa, but when they have associated dysplasia, higher rates of proliferation activity (Ki67) and beta-catenin positivity are found 4. In normal gastric mucosa there is a normal expression of Ki-67 in the proliferative zone near the neck region. This marker was used in one study along with p21, a cyclin-dependent kinase inhibitor that is regulated by p53 and normally confined to the epithelium in the upper foveolar component and surface epithelium, but not in the proliferative neck regions. Abnormal expression of Ki-67 and p21 has been reported in FGPs with dysplasia, as 100% (8 of 8) of them inverted their normal topographic distribution in the gastric mucosa in the presence of dysplasia 3, 7. Similar to this report, the immunohistochemical stain for Ki-67 demonstrated, in the areas of dysplasia in our case, an increased epithelial proliferation observed at the surface of the polyp and extending down to the foveolar region, contrasting to the normal restricted proliferative zone of the neck of the glands.
REFERENCES
- Abraham, S. C., B. Nobukawa, et al. (2000). "Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic adenomatous polyposis coli gene alterations." Am J Pathol 157(3): 747-54.
- Jalving, M., J. J. Koornstra, et al. (2003). "Dysplasia in fundic gland polyps is associated with nuclear beta-catenin expression and relatively high cell turnover rates." Scand J Gastroenterol 38(9): 916-22.
- Wu, T. T., S. Kornacki, et al. (1998). "Dysplasia and dysregulation of proliferation in foveolar and surface epithelia of fundic gland polyps from patients with familial adenomatous polyposis." Am J Surg Pathol 22(3): 293-8.
- Jalving, M., J. J. Koornstra, et al. (2003). "High-grade dysplasia in sporadic fundic gland polyps: a case report and review of the literature." Eur J Gastroenterol Hepatol 15(11): 1229-33.
- Abraham, S. C., S. J. Park, et al. (2002). "Sporadic fundic gland polyps with epithelial dysplasia : evidence for preferential targeting for mutations in the adenomatous polyposis coli gene." Am J Pathol 161(5): 1735-42.
- Dihlmann, S. and M. von Knebel Doeberitz (2005). "Wnt/beta-catenin-pathway as a molecular target for future anti-cancer therapeutics." Int J Cancer 113(4): 515-24.
- Hassan, A., L. M. Yerian, et al. (2004). "Immunohistochemical evaluation of adenomatous polyposis coli, beta-catenin, c-Myc, cyclin D1, p53, and retinoblastoma protein expression in syndromic and sporadic fundic gland polyps." Hum Pathol 35(3): 328-34.
- Gencosmanoglu, R., E. Sen-Oran, et al. (2003). "Gastric polypoid lesions: analysis of 150 endoscopic polypectomy specimens from 91 patients." World J Gastroenterol 9(10): 2236-9.
- Sekine, S., T. Shimoda, et al. (2004). "High-grade dysplasia associated with fundic gland polyposis in a familial adenomatous polyposis patient, with special reference to APC mutation profiles." Mod Pathol 17(11): 1421-6.
- Stolte, M., M. Vieth, et al. (2003). "High-grade dysplasia in sporadic fundic gland polyps: clinically relevant or not?" Eur J Gastroenterol Hepatol 15(11): 1153-6.
- Leggett, B. A., J. P. Young, et al. (1997). "Severe upper gastrointestinal polyposis associated with sparse colonic polyposis in a familial adenomatous polyposis family with an APC mutation at codon 1520." Gut 41(4): 518-21.
 Contributed by Rosemary A Recavarren, MD, Robert Schoen, MD, and Antonia R. Sepulveda, MD, PhD
Contributed by Rosemary A Recavarren, MD, Robert Schoen, MD, and Antonia R. Sepulveda, MD, PhD




) Syndromic FGPs arise through somatic mutations in the second allele of the adenomatous polyposis coli (APC) gene, whereas sporadic FGPs are caused by beta-catenin gene mutations. Although they are two different genes (APC and beta-catenin), they act through a common pathway, the Wnt-APC-beta-catenin pathway (Figure 5). Genetic mutations in these genes lead to deregulation in the activation of the Wnt-APC-beta-catenin pathway, which is responsible for both the syndromic and the sporadic FGPs.
Syndromic FGPs arise through somatic mutations in the second allele of the adenomatous polyposis coli (APC) gene, whereas sporadic FGPs are caused by beta-catenin gene mutations. Although they are two different genes (APC and beta-catenin), they act through a common pathway, the Wnt-APC-beta-catenin pathway (Figure 5). Genetic mutations in these genes lead to deregulation in the activation of the Wnt-APC-beta-catenin pathway, which is responsible for both the syndromic and the sporadic FGPs.