FINAL DIAGNOSIS
Clonal neoplasm, favor metastatic carcinoma
DISCUSSION
The findings were discussed with the patient's endocrinologist, who reported a prior history of renal cell carcinoma (RCC) with adrenal invasion and lymph node metastasis, and added that the clinical differential diagnoses included metastatic RCC versus a primary thyroid neoplasm.
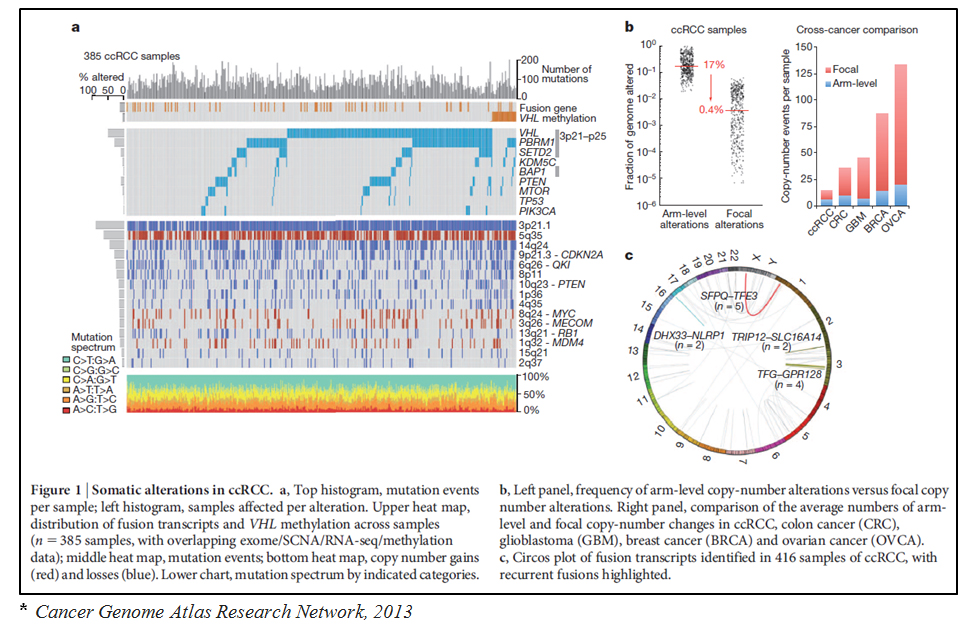
Renal cell carcinoma (RCC) accounts for 2-3% of adult malignancies, of which clear cell RCC (CC-RCC) comprises for 60-70% of cases. CC-RCC is a malignant renal tumor that arises from the epithelial cells of the proximal tubules. The classic morphology consists of compact nests of cells with clear cytoplasm, separated by a network of delicate vasculature. Tumor cells are typically positive for PAX2, PAX8, CD10, CAIX, vimentin, and broad spectrum cytokeratin, but are negative for CK7, CK20, CD117, and AMACR (P504S). CC-RCC is sporadic in ~95% of cases, and typically present in adults greater than 40 years of age. However, approximately 5% of cases are associated with von Hippel-Lindau (VHL) disease and show germline mutations in the VHL gene (3p25.3).
Wild-type VHL protein is a tumor suppressor that binds to the E3 ubiquitin ligase complex, and leads to degradation of the transcription factor Hypoxia-inducible factor 1-alpha (HIF1α). CC-RCC is typically characterized by biallelic inactivation of VHL, the loss of which leads to stabilization of HIF1α and increased expression of genes associated with angiogenesis and cellular proliferation (VEGFA, PDGFB, TGFA, EGFR, IGFBP3, SLC2A1, EPO, and CXCR4).
Sporadic Disease
Sporadic CC-RCC shows biallelic inactivation of VHL in ~90% of cases, which may occur through a combination of gene mutation, deletion, and/or DNA methylation. Loss of at least 1 copy of chromosome 3p is seen in >90% of sporadic tumors. Inactivating mutations in VHL are seen in ~50-60% of cases, and appear to be mutually exclusive with epigenetic inactivation of VHL through promoter methylation (~20% of cases). Cases that are VHL wild-type (<10% of CC-RCC), may show biallelic inactivation of TCEB1, a gene which encodes the elongin C component of the E3 ubiquitin ligase complex.
Additional mutations in other genes on chromosome 3p, most of which are involved in chromatin remodelling or histone modification, are also seen (PBRM1: 40-45%; BAP1:10-15%; SETD2: ~12%). Mutations in the PIK3K-AKT-mTOR pathway are seen in 20-30% of sporadic CC-RCC, and mutations in tumor suppressors and other cell cycle regulators are seen in ~40% of cases.
Additional chromosomal alterations are frequently seen, and include gains of 5q and 7q, losses of 8p ± 8q, MYC, MDM4, or loss of heterozygosity (LOH) of 9p,14q (including HIF1α), and 18q. Copy number loss of 4p, 14q, and 9p are associated with more aggressive disease.
Additional epigenetic changes seen in sporadic CC-RCC include methylation of other genes located in chromosome 3p (RASSF1, FAM107A, DLEC1) as well as FHIT and Wnt pathway inhibitors (SFRP1, SFRP4, SFRP5, DKK1, DKK2, and DKK3).
Von Hippel-Lindau disease
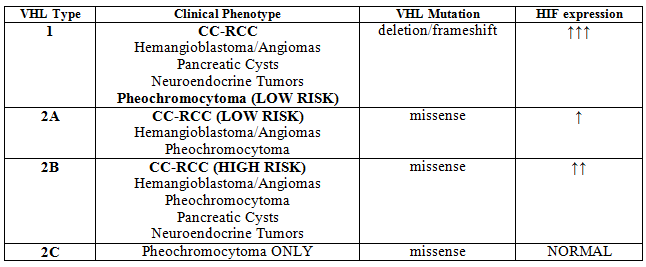
Von Hippel-Lindau (VHL) disease is an autosomal dominant (AD) familial tumor predisposition syndrome with germline inactivating mutations or deletions in the VHL gene. Loss of function of the other allele by a second genetic hit results in disease expression. The disease occurs in 1 of 36,000 live births and is characterized by the development of multiple tumor types. In VHL disease patients who develop CC-RCC, these tumors are frequently multiple and bilateral. Other tumors that frequently occur as part of this syndrome include retinal angiomas, CNS hemangioblastomas, pancreatic serous cystadenomas, and papillary cystadenomas of the epididymis. Less common tumors include adrenal or extra-adrenal pheochromocytomas, pancreatic neuroendocrine tumors (typically non-functional), endolymphatic sac tumors, and rarely head & neck paragangliomas.
VHL disease can be divided into 4 phenotypic categories:
REFERENCES
![]() Contributed by Siddharth Bhattacharyya, MD; Simion Chiosea, MD
Contributed by Siddharth Bhattacharyya, MD; Simion Chiosea, MD