DIAGNOSIS
Medium Chain Acylcarnitine Dehydrogenase Deficiency
DISCUSSION
Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) was first described in 1976. It is a recessive disease and is the most common defect in mitochondrial B-oxidation in humans occurring at a rate of 1/15,000-1/30,000 [1]. Undiagnosed MCADD has considerable early mortality due to impaired ketogenesis during periods of glycogen depletion. If unrecognized, this can lead to hypoketotic hypoglycemia causing coma and death in 20-25% of patients. Forty percent of individuals who survive the coma suffer some degree of neurological disability.
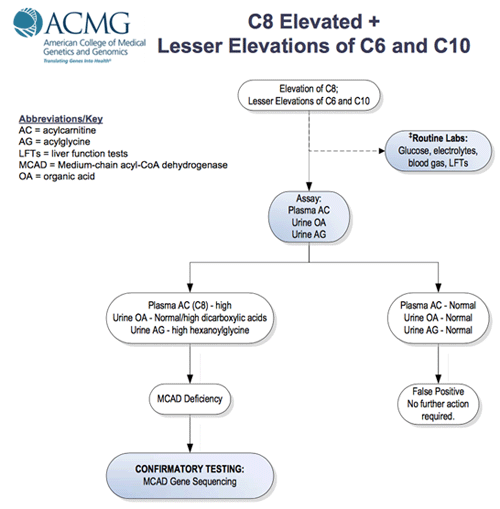
The newborn screen (NBS) identifies MCADD, and other metabolic deficiencies, with an acylcarnitine profile. The finding of elevated C8 as well as C6 and C10 is diagnostic for the disease but confirmation is required (Figure 3) .
Assuming the premature birth rate in the United States is 9.6% and the rate of MCADD is 1/15,000-1/30,000, (assuming the exclusivity of each variable) the rate of this event will be between approximately 1/150,000 and 1/300,000 live births making this an uncommon occurrence, however, a finding of which NICU personnel should be aware.
Medium chain fatty acids make up less than 12% of fat in human milk; however, they account for 40% of the fat in premature formulas. Medium chain fatty acids are believed to have better absorption than cows' milk alternatives. Specifically, Similac Special Care and Similac Fortifier (used in this patient) both list the medium chain triglycerides as there forth ingredient. The surprisingly large buildup of medium chain acylcarnitine esters in our patient owed to the inability to catabolize medium chain fatty acids that were being provided in excess.
Luckily for this child and for other children born with MCADD, the NBS identifies patients with MCADD early which allows for the quick recognition of the disease and the subsequent dietary management. The infant was always on either dextrose water or Similac Care/breast milk and Similac Fortifier. Since the second and third ingredients in Similac premature formulas are non-fat milk and corn syrup respectively, there was not an immediate danger of hypoglycemia.
Follow-up genetic testing identified the most common mutation c.985A>G (p.Lys329Glu) as well as a less common mutation c.347G>A (p.Cys116Tyr).
REFERENCES
![]() Contributed by Nicholas Barasch MD MS and Steven Dobrowolski PhD
Contributed by Nicholas Barasch MD MS and Steven Dobrowolski PhD