FINAL DIAGNOSIS
The diagnosis of extensive cerebral venous sinus thrombosis secondary to hypofibrinolysis due to the homozygous PAI-1 4G/4G polymorphism and elevated PAI-1 antigen and activity levels was confirmed.
DISCUSSION
Fibrinolysis
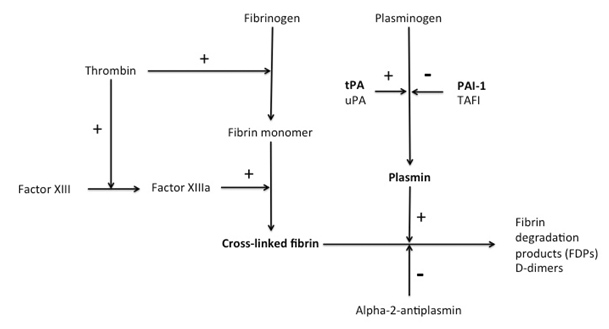
The fibrinolytic cascade is a complex system of serine proteases, their inhibitors, activators and receptors, which act to regulate the activation of plasminogen to plasmin.(2) The final steps in the coagulation cascade lead to thrombus formation by conversion of fibrinogen to fibrin monomers and covalent cross-linking of fibrin strands to form cross-linked fibrin (Figure 3).(3)
Figure 3: Activators and inhibitors of the fibrinolytic system. tPA - tissue plasminogen activator, uPA - urokinase plasminogen activator, PAI-1 - plasminogen activator inhibitor-1, TAFI - thrombin activatable fibrinolysis inhibitor.
Plasminogen is a circulating plasma zymogen, which can be converted either on cell surfaces or on the surface of a thrombus to active plasmin by the action of two activators: tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA).(2, 4) While tPA is mainly involved in the dissolution of a thrombus, the major role of uPA is in pericellular proteolysis during tissue remodeling.(5) The catalytic activity of tPA is greatly enhanced when it is complexed with plasminogen on lysine-binding sites of fibrin.(2) Plasmin is the major fibrinolytic protease, which degrades cross-linked fibrin and fibrinogen into soluble fibrin degradation products, D-dimers and fibrinogen degradation products.(6) In this regard, fibrin is both a product of the coagulation cascade and a substrate for the fibrinolytic cascade (Figure 3). Upon cleavage of fibrin, additional C-terminal lysine residues are exposed, which creates positive feedback for the activation of plasminogen.(2)
Inhibitors of fibrinolysis can be divided into three main categories: plasmin inhibitors, plasminogen activator inhibitors and attenuators.(2) The main inhibitors of plasmin are alpha-2-antiplasmin, alpha-2-macroglobulin and protease nexin, of which alpha-2-antiplasmin is the most important. Alpha-2-antiplasmin is a serine protease inhibitor or serpin that binds in a 1:1 stoichiometry with the active site serine of plasmin, forming an irreversible complex.(7) Both molecules subsequently lose their catalytic activity and are cleared from the circulation. Both plasmin and alpha-2-antiplasmin have half-lives of approximately 50 hours and the plasma concentration of plasmin (0.2mg/ml) is about twice that of alpha-2-antiplasmin (0.07mg/ml).(8)
The most important class of fibrinolysis inhibitors is the plasminogen activator inhibitors. In order of reaction rate constants, these include plasminogen activator inhibitor-1 (PAI-1), PAI-2, protease nexin and PAI-3.(9) The most ubiquitous plasminogen activator inhibitor is PAI-1.
Thrombin-activatable fibrinolysis inhibitor (TAFI) is a fibrinolysis attenuator, which is activated to TAFIa by thrombin/thrombomodulin and by plasmin.(10, 11) TAFIa can cleave lysine residues from fibrin, thus decreasing the binding of plasminogen to fibrin and reducing the fibrinolysis-enhancing effect of these residues.(12)
Plasminogen Activator Inhibitor-1 (PAI-1)
PAI-1 is a 52kDa single-chain, labile, glycoprotein serpin that is produced by endothelial cells, platelets, megakaryocytes, monocytes, macrophages, hepatocytes and adipocytes.(13-17) PAI-1 is present at a concentration of 60 ng/ml in plasma, about 5-10 times higher than the concentration of tPA and 50 times higher than the concentration of uPA.(8) Platelet PAI-1 accounts for approximately half of circulating PAI-1 activity but is less active than plasma PAI-1.(18) PAI-1 is also an acute phase reactant, likely resulting from increased hepatocyte production.(19) The plasma half-life of PAI-1 has been reported to be approximately 5-7 minutes (8) and the active form of PAI-1 is stabilized in the circulation by noncovalent binding to the glycoprotein vitronectin.(20) Four different conformations of PAI-1 have been described: 1) the active form that reacts with plasminogen activator; 2) a latent form that is nonreactive but can be converted to the active form; 3) a substrate form that can be cleaved by plasminogen activators but is noninhibitory; and 4) the inert form of PAI-1 generated by the cleavage of the reactive site.(21)
PAI-1 inhibits both tPA (single-chain and two-chain) and uPA (two-chain only) by forming a stable 1:1 complex causing loss of activity.(22) Elevations in PAI-1 levels can lead to hypofibrinolysis. Increased plasma levels of PAI-1 have been associated with both venous and arterial thrombosis.(23-25)
PAI-1 concentrations are higher in older patients, in men, in current/ former smokers, in obese patients, in patients with evidence of proteinuria or a chronic inflammatory state (elevated C-reactive protein) and in patients with the metabolic syndrome.(26) It is likely that the production of PAI-1 by adipose tissue, in particular by tissue from omentum, could be an important contributor to the elevated plasma PAI-1 levels observed in insulin resistant patients.(27)
The PAI-1 gene and 4G/5G polymorphism
The PAI-1 gene is located on chromosome 7 and is 12 kb long, consisting of 9 exons and 8 introns. The transcription start point is located 142 nucleotides upstream from the start codon.(28) The most frequently described polymorphism in the PAI-1 gene is the 4G/5G single base pair insertion/ deletion polymorphism (allele frequency 0.53/0.47), which is located 675 bp upstream of the transcriptional start site (Figure 4).(29) The 5G polymorphism is more common and allows a transcriptional repressor to bind to the transcriptional activator, thus reducing mRNA transcription and PAI-1 levels. The 4G polymorphism causes inhibition of binding of the transcriptional repressor, allowing unopposed action of the transcriptional activator and elevated PAI-1 levels.(30, 31)
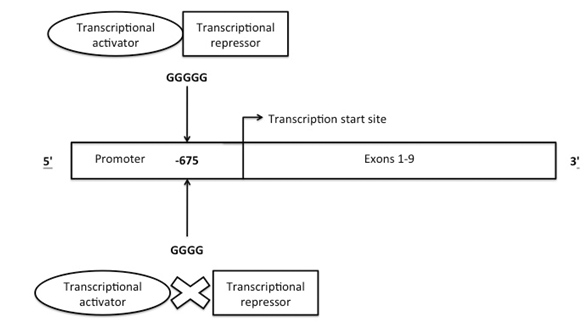
Figure 4: Structure of PAI-1 gene showing site of 4G/5G polymorphism 675bp upstream of the transcriptional start site and possible mechanism of transcriptional control.
The PAI-1 4G/4G polymorphism has been associated with an elevated risk of certain venous thromboembolic disorders. Sartori et al. found an association between the 4G/4G genotype and a greater risk of thrombosis both in symptomatic thrombophilia patients (OR 2.85, 95% CI 1.26-6.46) and in idiopathic deep venous thrombosis (DVT) patients (OR 3.1, 95% CI 1.26-7.59).(32) Balta et al. found an association between both the 4G/4G and the 4G/5G genotypes with an increased risk of internal organ thrombosis, especially portal vein thrombosis.(33) Segui et al. studied 190 unrelated patients with DVT and 152 healthy controls and found no significant difference in allele frequencies between the groups. The authors reported that, in the DVT group, PAI-1 antigen levels were influenced by the 4G allele dosage, triglyceride levels and body mass index (BMI).(34) The 4G/4G genotype has also been associated with pregnancy complications due to placental insufficiency (35, 36) and with myocardial infarction.(37, 38) In all of the above studies, the PAI-1 4G/4G genotype was associated with elevated PAI-1 activity and/ or antigen levels.
The evidence for the association between PAI-1 4G/5G genotype and cerebral sinus thrombosis is conflicting. While several case studies have highlighted a possible relationship between the 4G allele and cerebral sinus thrombosis (39-41), case-control studies to date have failed to find a significant independent relationship.(33, 42) One study looking at PAI-1 4G/5G genotypes in carriers of the factor V Leiden (FVL) mutation did find that the concurrence of FVL and homozygosity for the 4G allele lead to an increased risk for cerebral sinus thrombosis, supporting the assumption that in carriers of the FVL mutation a further prothrombotic factor may be necessary for the development of a manifest thrombotic event.(43)
SUMMARY
This case reports on a 22 year old obese female with no acquired thrombophilic risk factors, who was diagnosed with extensive cerebral sinus thrombosis. A detailed thrombophilia workup demonstrated persistently elevated PAI-1 activity levels, with an elevated PAI-1 antigen concentration and homozygosity for the PAI-1 4G allele (4G/4G genotype). The patient was treated with indefinite warfarin anticoagulation due to the unprovoked nature of her thrombotic event. A follow-up brain MRI and angiogram performed five months later showed patent cerebral venous sinuses and normal venous drainage. However, the patient was left with permanent vision loss in her left eye and only partial visual acuity in her right eye.
Disturbances in the fibrinolytic system, in particular PAI-1, have been related to an increased risk of both arterial and venous thrombosis. There also appears to be a relationship between PAI-1 levels and obesity, the metabolic syndrome and insulin resistance. While the evidence for a link between the 4G/4G genotype and cerebral sinus thrombosis is weak, we were unable to find any other inherited or acquired cause of this patient's thrombotic event. PAI-1 gene analysis and PAI-1 activity are not considered to be standard tests for evaluation of patients with thrombosis. However, in young patients with an otherwise unexplained thrombotic event additional investigation including evaluation of fibrinolytic system should be considered.
REFERENCES
![]() Contributed by Jansen N Seheult, Mike Meyers, Irina Chibisov
Contributed by Jansen N Seheult, Mike Meyers, Irina Chibisov