DIAGNOSIS
Type 1 autoimmune pancreatitis
INTRODUCTION
Autoimmune pancreatitis is a rare fibroinflammatory disease that accounts for 5-6% of all cases of chronic pancreatitis1. Due to its non-specific clinical presentation, laboratory characteristics, and radiological findings autoimmune pancreatitis can be a challenging diagnosis for clinicians especially since one of the main differential diagnoses is pancreatic adenocarcinoma1. Autoimmune pancreatitis is highly responsive to steroid treatment, making an accurate diagnosis essential.
Currently there are two subtypes of autoimmune pancreatitis. Type 1 is associated with elevated serum IgG4 levels and tissue invasion by plasma cells positive for IgG4. Histologically type 1 autoimmune pancreatitis is characterized by lymphoplasmacytic infiltration, storiform fibrosis, obliterative phlebitis, and elevated IgG4-positive plasma cells within the tissue. Type 2 autoimmune pancreatitis is not associated with IgG4 and is histologically characterized by neutrophilic infiltration into the pancreatic duct epithelium. Type 2 disease is also associated with inflammatory bowel disease, while type 1 is not. Type 1 autoimmune pancreatitis is more prevalent than type 210. The focus of this case report will be type 1 autoimmune pancreatitis.
The first presumed case of what is now called type 1 autoimmune pancreatitis was reported in 1961 by Sarles and colleagues when they described a case of pancreatitis associated with hypergammaglobulinemia and suggested autoimmunity as a potential mechanism3. In 1995, Yoshida and colleagues used the term autoimmune pancreatitis to describe patients with characteristic laboratory, clinical, and histological findings1,5. Before this, type 1 autoimmune pancreatitis had several names based on its clinical and morphological appearance. These names include lymphoplasmacytic sclerosing pancreatitis, chronic sclerosing pancreatitis, and non-alcoholic duct destructive chronic pancreatitis1.
PATHOGENESIS
The pathophysiological mechanism for type 1 autoimmune pancreatitis is currently unknown. It is frequently associated with other autoimmune diseases including rheumatoid arthritis and Sjogrens disease. Patients with type 1 autoimmune pancreatitis often have an increased number of activated CD4-, CD8+ T-cells with increased HLA-DR in the peripheral blood and pancreas5.
CLINICAL PRESENTATION
Type 1 autoimmune pancreatitis usually affects males over the age of 501, but the clinical presentation can vary. The most common symptoms are painless jaundice and abdominal pain. Additional symptoms can include weight loss, diabetes, and in rare cases severe abdominal pain and acute pancreatitis. Because this disease can be systemic, patients may also present with extra-pancreatic manifestations, such as renal insufficiency, and pulmonary infiltrates1.
DIAGNOSIS
The diagnosis of type 1 autoimmune pancreatitis spans across several medical disciplines. Classic radiological findings include diffuse pancreatic ("sausage-shaped") enlargement with homogenous attenuation and peripheral enhancement of the pancreas along with diffuse narrowing of the pancreatic duct. Focal involvement most often involves the head of the pancreas, which often mimics pancreatic adenocarcinoma1. A CT can often reveal lymphadenopathy of the cervical, abdominal, or hilar lymph nodes5.
Type 1 autoimmune pancreatitis is associated with other autoimmune diseases including rheumatoid arthritis and Sjogren disease, making laboratory testing and monitoring imperative in suspected patients. Individuals may have elevated titers of immunogammaglobulins, antinuclear antibody, rheumatoid factor, and/or anticarbonic anhydrase antibodies2. One of the most studied and possibly controversial lab values affiliated with type 1 autoimmune pancreatitis is serum IgG4 levels. Hamano and colleagues found that serum IgG4 concentrations of 135 mg/dL had a sensitivity of 95% and a specificity of 97% when diagnosing type 1 autoimmune pancreatitis versus pancreatic cancer7. However, Ghazale et al found the sensitivity of serum IgG4 levels to be only around 77%. In the same study, patients with serum IgG4 levels greater than 280 mg/dL (twice the normal value) had a greater than 95% likelihood of having type 1 autoimmune pancreatitis. Their study also illustrated that serum IgG4 levels had a high negative predictive value2. The diagnostic utility of serum IgG4 levels is also controversial because an elevated serum IgG4 levels is non-specific and can be found in a number of other illnesses including atopic dermatitis, asthma, parasitic diseases, pemphigus, and pancreatic carcinoma3.
On gross examination the pancreas can be firm and indurated1, often with a thick capsule5. A focal mass may be present1, but often without calculi, pseudocysts or necrosis, which are common in alcoholic-associated chronic pancreatitis4. Microscopically, the hallmark finding of type 1 autoimmune pancreatitis is a periductal infiltrate composed of plasma cells and lymphocytes1. This infiltrate is closely associated with fibrosis of pancreatic parenchyma1. The lymphocytic component of the inflammation is mainly composed of CD4+, CD8+ T lymphocytes and few B cells3. The acini are often atrophic3, and the interlobular septa are often thickened by the lymphoplasmacytic infiltrate and a proliferation of myofibroblasts1. Perineural inflammation has been reported5. Other characteristic findings include the presence of obliterative venulitis that spares the arterioles2.
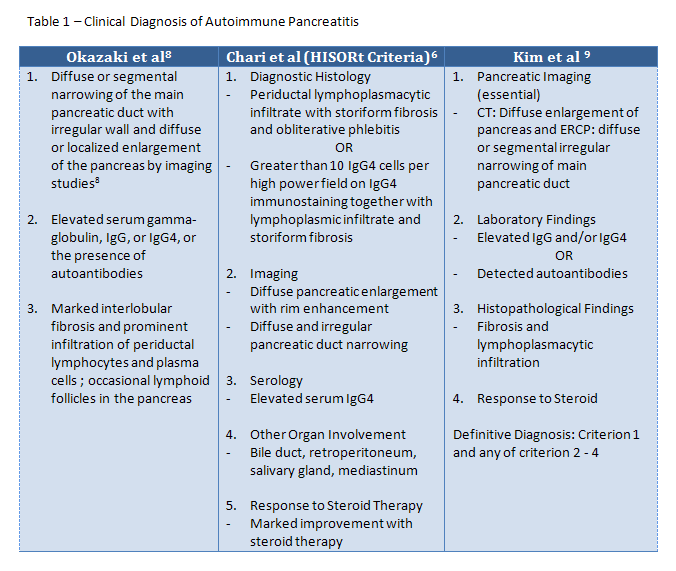
Several groups have developed criteria for the diagnosis of type 1 autoimmune pancreatitis. These criteria can be found in Table 1. Kamisawa et al was the first group to devise diagnostic criteria in 20025. These criteria were later revised into the one proposed by Okazaki et al in 20068. This group of criteria focuses on the imaging characteristics as well as the histopathological and serological findings. The HISORt criteria was developed by Chari et al at the Mayo Clinic and encompasses histologic, imaging, serologic, extrapancreatic involvement, and steroid therapy response characteristics to the diagnosis of type 1 autoimmune pancreatitis6. The criteria developed by Kim et al is similar to the HISORt criteria except it does not take into account the extrapancreatic manifestations of the disease, and imaging characteristics of the pancreas is necessary for diagnosis. Across criteria, the microscopic and imaging requirements are very similar.
DIFFERENTIAL DIAGNOSIS
The most important differential diagnosis for type 1 autoimmune pancreatitis is pancreatic adenocarcinoma. Erroneous diagnoses of pancreatic adenocarcinoma account for 2.5% of all pancreaticoduodenectomies performed on autoimmune pancreatitis patients5. Pancreatic carcinoma shares many qualities with type 1 autoimmune pancreatitis which include an elderly preponderance, new onset diabetes mellitus, painless jaundice, focal swelling of the pancreas, strictures in the pancreatic and biliary ducts, and elevated tumor markers5. Alcohol-associated chronic pancreatitis can also be included in the differential, but can be differentiated from autoimmune pancreatitis based on characteristics previously described.
TREATMENT
The foundation treatment for type 1 autoimmune pancreatitis is corticosteroids. Steroid regimens usually begin with a week of prednisone 40 mg daily followed by tapering of the dose by 5 mg per week. Prior to and during treatment, a patient should undergo imaging and lab testing in order to monitor response1. Steroid response consists of the pancreas returning to normal size and a normalizing of pancreatic function5. Patients with discrete masses undergoing corticosteroid therapy should have complete resolution of the mass as determined by imaging. Carcinoma of the pancreas can have some response to steroids, but complete resolution of the mass will not occur. Patients who fail to respond to steroids should have a surgical biopsy1.
CONCLUSION
Type 1 autoimmune pancreatitis is a challenging diagnosis for clinicians and pathologists alike and an accurate diagnosis is crucial to avoiding unnecessary and often life-changing surgical procedures. The wide variety of presentations, the lack of a highly sensitive and specific test, and the inaccessibility of the pancreas add to the difficulty of achieving an accurate diagnosis. Using multiple modalities across medical disciplines to screen patients suspicious for type 1 autoimmune pancreatitis can increase the likelihood of reaching an accurate diagnosis, which will provide the best possible outcome for the patient.
REFERENCES
![]() Contributed by Russell Silowash, DO and Aatur Singhi, MD, PhD
Contributed by Russell Silowash, DO and Aatur Singhi, MD, PhD