
![]() Contributed by Sarah K Harm, MD and Jeffrey A Kant, MD, PhD
Contributed by Sarah K Harm, MD and Jeffrey A Kant, MD, PhD
PATIENT HISTORY
The patient is a nine month old Caucasian boy who, as a newborn, failed to pass meconium prior to his 48 hour discharge from the hospital. He was the product of an uncomplicated, term pregnancy. He went on to develop chronic constipation and was clinically diagnosed with Hirschsprung disease. Biopsies were performed and the clinical diagnosis of Hirschsprung disease was pathologically confirmed by intestinal aganlionosis involving only the rectosigmoid colon (i.e. short-segment disease). The patient had no other congenital anomalies and there was no family history of Hirschsprung disease so he was therefore classified as a case of isolated Hirschsprung disease.
What genetic testing, if any, would you perform first to support the diagnosis of Hirschsprung disease?
ANSWER
RET gene testing (exons 2, 3, 5, 6, 9, 10, 12, 13 & 17)
EXPLANATION
Inactivating mutations of the RET gene, located on chromosome 10q11.2, are associated with Hirschsprung disease. The RET gene encodes a receptor tyrosine kinase expressed primarily in neural crest and urogenital precursor cells. [1] Disease-associated mutations have been identified scattered throughout the gene; however, most commonly are found in exons 2, 3, 5, 6, 9, 10, 12, 13 & 17. [2,3] Most mutations are missense or nonsense codon changes. A few splice-site mutations have been reported as well as large and small deletions.
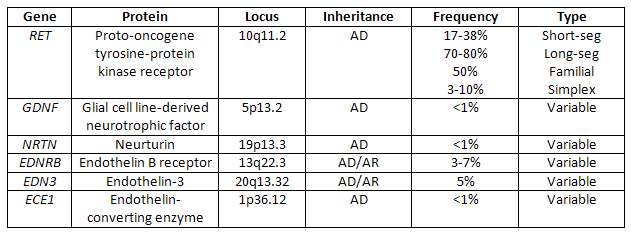
Additional genes associated with isolated Hirschsprung disease have been identified but occur with frequencies much less than that of the RET gene [Table 1]. Although RET is the primary gene underlying Hirschsprung disease, particularly in long-segment disease, RET mutations show incomplete, sex-dependent penetrance and do not always result in the Hirschsprung phenotype. [2,3] Importantly, a negative RET mutation analysis does not rule out an underlying genetic etiology.
Table 1: Genes associated with isolated Hirschsprung disease. Adapted from GeneReviews. [4]
While Hirschsprung disease is associated with inactivating mutations in the RET proto-oncogene, gain-of-function mutations (mostly missense mutations) are associated with Multiple Endocrine Neoplasia Type 2A/B (MEN2A/B) syndromes. Exons 10, 11, 13 & 14 of the RET gene are implicated in MEN2A while mutations in exons 15 & 16 are found in MEN2B. While overlap exists between the exons involved with MEN2A and Hirschsprung disease, a clear association between these disease entities has not been found. There are a few case series however, that have found associations ranging from 1-5% of the individuals tested. [1,5-7] The conclusions support the incomplete penetrance of Hirschsprung disease and the difficulty of using genetic testing to predict disease phenotype.
The utility of genetic testing in the face of biopsy proven Hirschsprung disease is questionable. Current recommendations include genetic testing of RET and, if negative, EDN3 and/or EDNRB if a monogenic nonsyndromic case is likely and/or confirmed. [4] Mutation positive Hirschsprung disease patients are clinically treated identical to mutation negative cases, albeit with additional genetic counseling once the patient is of childbearing age.
REFERENCES