FINAL DIAGNOSIS Fabry Disease - Mosaic pattern
Genetic analysis of the patient reveals that she carries the familial mutation R112H in the GLA gene. The patchy distribution of podocyte involvement on light microscopy and mild proteinuria clinically suggest a mosaic pattern and heterozygous genotype in this female patient.
DISCUSSION
Background
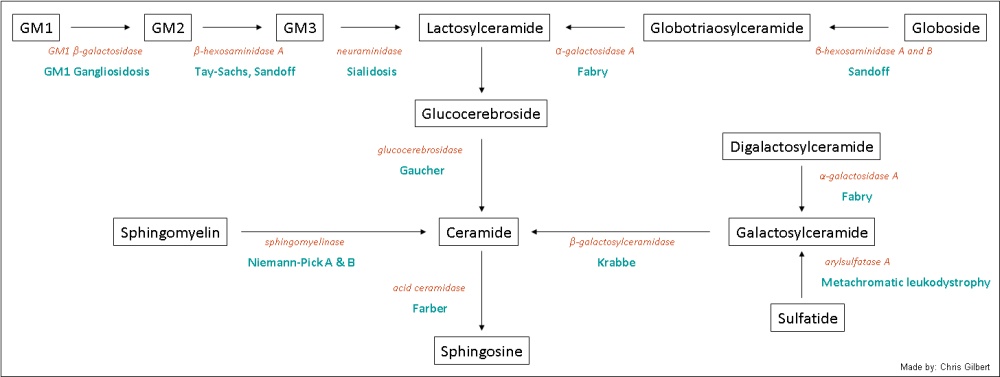
Fabry disease (also known as Fabry's disease, Anderson-Fabry disease, angiokeratoma corporis diffusum and alpha-galactosidase A deficiency), first described by Drs. Johann Fabry and William Anderson in 1898, is a lysosomal storage disorder with a recessive, X-linked, Mendelian genetic inheritance pattern (1). It is caused by one or more of hundreds of characterized mutations in the GLA gene (2). GLA, short for "galactosidase, alpha," a 12-kb gene mapped to the long arm (Xq22.1) of the X chromosome, encodes the enzyme alpha galactosidase A, which plays an important role in glycosphingolipid metabolism: specifically the hydrolysis of the terminal galactose in alpha-galactosyl moieties. A mutation in this pathway results in the accumulation of galactosylceramide and globotriaosylceramide in lysosomes throughout the body causing multi-organ disease and dysfunction.
Natural History
In males, the natural history of the disease can be conceptually divided into three sequential time periods. The first manifestations of the disease include acroparesthesias, hypohydrosis, nausea, post-prandial diarrhea and corneal opacities that occur with an average onset of 5 years of age (3). "Fabry crises," which often occur in childhood, are described as burning or tingling of the limbs triggered by environmental stressors without swelling, erythema or tenderness. The non-specific nature of these complaints and absence of physical findings makes diagnosis exceedingly difficult. In the second and third decades, accumulation of abnormal glycosphingolipid in the heart, kidney, and brain begin to figure prominently. It is at this point that renal hyperfiltration, microalbuminuria, and proteinuria are observed. Likewise, in the heart, hypertrophic cardiomyopathy, valvular disease, and mitral insufficiency are common findings. Neurological function is compromised including significant hearing loss, tinnitus, and loss of touch sensation. On the skin, angiokeratomas are frequently identified. With advanced age, these disease manifestations continue to take their course. In the heart, electrocardiogram abnormalities including arrhythmias are of major concern. In the brain, the risk for transient ischemic attacks and strokes in men 25-44 years of age is 12 times higher than the general population (4). Renal insufficiency leading to kidney failure frequently occurs.
X-inactivation in female patients makes the presentation and progression of heterozygous carriers highly unpredictable. As little as 5-10% of galactosidase activity is required to (5) prevent clinically significant accumulations. Therefore the natural history of afflicted female patients varies significantly from those that resemble men, to patients, such as the one from this case, who live a long life with much slower disease progression.
Epidemiology
Worldwide, the incidence of Fabry disease ranges from 40,000 to 117,000 (6). There is no known ethnic predilection. Males are disproportionately affected and usually diagnosed in early adulthood while a significant proportion of female patients present later or go undiagnosed. Untreated men see their life expectancy reduced by roughly 20 years compared to the general population (3). Women on average see a reduction of just 15 years (7). These statistics are rapidly changing due to the availability of enzyme replacement therapy.
Diagnosis
The gold standard for the diagnosis of Fabry disease in men requires the measurement of depressed alpha-galactosidase A activity in peripheral leucocytes and/or alpha galactosidase A gene sequencing with the identification of a disease-causing mutation. In women, enzyme activity levels are notoriously unreliable and gene sequencing testing is the only acceptable method.
In the kidney, the pathologic findings are distinctive. On light microscopy, the glomeruli contain prominent, foamy, vacuolated, PAS-negative inclusions in the podcytes, mesangial cells and endothelial cells. These inclusions are birefringent and autofluoresce with polarized illumination. They can be well visualized on toluidine blue staining. Electron microscopy shows scattered and patchy to diffuse involvement of the renal glomeruli by numerous lamellated, myeloid or "zebra" bodies. It must be noted that these inclusions are not unique to Fabry disease and are found in other lysosomal storage disorders. Progression of disease results in fusion of foot processes, focal glomerular necrosis and thickening of glomerular and tubular basement membranes (8).
Recognition of these characteristic findings can be complicated in instances of coexisting disease and/or severe renal insufficiency changes.
Treatment
The availability of enzyme replacement therapy in the form of Replagal (available in Europe) and Fabrazyme (available in the U.S.) have dramatically changed the natural history and medical management of these patients. Formerly, treatment consisted primarily of symptom management. But enzyme replacement therapy is not a panacea, with reports of treatment failure secondary to alloimmunization to the exogenous enzymes (3). Close medical management including frequent laboratory studies, echocardiography, and renal evaluation are still recommended.
REFERENCES & ADDITIONAL RESOURCES
![]() Contributed by Chris Gilbert, MD and Sheldon Bastacky, MD.
Contributed by Chris Gilbert, MD and Sheldon Bastacky, MD.