FINAL DIAGNOSIS
Angioimmunoblastic T-cell lymphoma (AITL).
DISCUSSION
AITL is a peripheral T-cell lymphoma characterized by systemic disease, a polymorphous infiltrate in lymph nodes, along with a prominent proliferation of follicular dendritic cells (FDC) and high endothelial venules (HEV).1 The diagnosis of this disease is based on a constellation of clinical, morphologic, immunophenotypic, cytogenetic and molecular evidence. It tends to affect middle aged and elderly population with equal sex predilection.1,2 T-cell lymphomas in general and AITL in particular have varied clinical and histologic features. This is complicated by the fact that they tend to retain some of the functional characteristics of non-neoplastic T-cells (production of cytokines and immune cell stimulation) and present with an obscuring population of a variety of non-neoplastic cells.1,3 The inappropriate and non-specific immune cell stimulation is associated with a paradoxical combination of hyperactivity of the immune system and immunosuppresion.2 Patients present at an advanced stage of disease due to frequent delay in correct diagnosis as clinical and pathological features of AITL overlaps many other reactive and neoplastic processes.1,4 Typical clinical presentation is characterized by fatigue, weight loss, pruritic skin rash, fever, hepatosplenomegaly, and generalized lymphadenopathy. Hematological features may include autoimmune hemolytic anemia and varying degree of thrombocytopenia.1,2,4 Polyclonal hypergammaglobunemia can be seen in some cases secondary to B-cell recruitment and activation by neoplastic T-cells.1,2 Patients are prone to opportunistic infections which appear to be the major cause of mortality and morbidity.2 These clinical features although classical for AITL, are not specific.
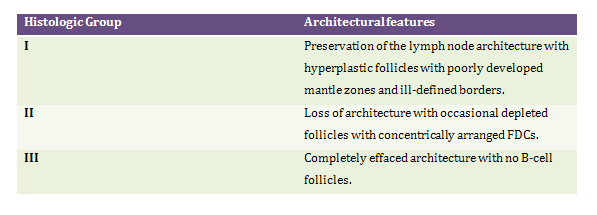
Microscopic examination is characterized by partial effacement of lymph node architecture with perinodal extension and sparing of cortical sinuses. There is marked expansion of the paracortex with proliferation of arborizing HEV. In most instances lymphoid follicles are regressively transformed however hyperplastic follicles have been documented. The neoplastic cells form small clusters around follicles and HEVs. The paracortical polymorphous infiltrate is comprised of small to intermediate sized lymphocytes with clear to pale cytoplasm, distinct cytoplasmic membrane and minimal cytologic atypia. Admixed in this infiltrate are seen plasma cells, eosinophils, histiocytes and proliferating FDCs. Histologic features of some perhaps early cases of AITL show expansion of paracortex with prominent hyperplastic follicles mimicking reactive hyperplasia.1-4 Based on architectural features AITLs have been categorized into 3 overlapping groups4:
Based on the above histologic features the morphological differential diagnosis include1-3
Immunophenotypically neoplastic T-cells express most pan T cell antigens (CD2, CD3 and CD5), CD4 and the follicular T-helper cell (FTHC) markers including CD10, CXCL13 and PD-1.1,2,4,5 CD8 positive reactive T-cells can be seen admixed in the infiltrate. There is characteristic expansion of FDC meshwork, as evidenced by immunoexpression of CD21, CD23 and CD35, around the HEVs.1 AITLs are almost always associated with Epstein Barr Virus (EBV) positive polytypic B-immunoblasts, of varying size, admixed in the polymorphous infiltrate. EBV positive Reed-Sternberg like B cells may be present, mimicking Classic Hodgkin lymphoma.1 This characteristic immunophenotype, especially FTHC and FDC markers, help distinguishing AITL from other peripheral T-cells neoplasms which lack the expression of FDC and FTHC markers, Classic Hodgkin lymphoma in which the neoplastic cells mark with CD30 and CD15 and marginal zone B-cell lymphoma which marks with the usual B-cell markers. The EBV positive large cells in Hodgkin lymphoma are the neoplastic Reed-Sternberg cells (CD30 positive) unlike in AITL where the EBV positive large cells represent non-neoplastic activated B-cells.1,2
Based on the most recent immunophenotyping and gene profiling studies, the cell of origin is a CD4 positive follicular helper T-cells (FTHC).1,2,4,5 The recently studied markers by gene profiling, CXCL13 and PD1, have been shown to be expressed by FTHC and aids in distinguishing AITL from other T-cell neoplasms.1,2,4,5 CXCL13, a chemokine that is produced in high quantities by normal T-follicular helper cells following co-stimulation via the CD28 and T cell receptors.2 Neoplastic T-cells demonstrate cytoplasmic immunoexpression with a perinuclear dot.4 Programmed Cell Death-1 (PD-1) is a member of the CD28 receptor family that regulates the cellular immune response.4,5 It is also seen as cytoplasmic immunoexpression, however stronger in intensity than CXCL13. 4 Yu et al4 studied CXCL13 and PD-1 expression in AITL and other peripheral T-cell lymphomas and demonstrated that very few cases from the latter group showed focal and weak staining. The non neoplastic lymphoid tissue demonstrated reactivity for both the markers in a subset of CD4 positive T-cells in the germinal centers. These markers, in conjunction with CD10 and Bcl-6, have been demonstrated to be characteristic for AITL thus helping in distinguishing from other peripheral T cell lymphomas.2,4,5
75-90% of AITL show clonal rearrangement of T-cell receptor and 25-30% also shows clonal immunoglobulin gene rearrangement even in the absence of a co-existing overt B-cell neoplasm. The most frequent cytogenetic abnormalities encountered include trisomy 3, trisomy 5 and additional X chromosome. Comparative genomic hybridization studies have shown gains of 22q, 19 and 11q13 and losses of 13q in a subset of cases.1 Although these cytogenetic findings are non-specific, they help in excluding non-neoplastic reactive lymphoid conditions which lack these abnormalities.
AITLs usually have an aggressive clinical course with median survival of less than 3 years.1 Susceptibility to infections makes treatment options challenging. Supervening or recurrent EBV positive large B cell lymphomas can occur.1,2
The current case demonstrates a classic clinical picture with characteristic histologic and immunophenotypic findings. The morphologic and immunophenotypic features were helpful in confirming the neoplastic nature and expression of CD10, Bcl-6, CXCL13 and PD-1 was very helpful in excluding other peripheral T-cell lymphomas. This disease is a great mimicker and to correctly diagnose it requires a high index of suspicion and appropriate use of immunophenotyping studies especially utilizing the newer FTHC markers. Larger studies are needed to validate the use of CXCL13 and PD1 in routine practice.
REFERENCES
![]() Contributed by Somak Roy, MD and Fiona E. Craig, MD
Contributed by Somak Roy, MD and Fiona E. Craig, MD