
FINAL DIAGNOSIS HYPOCHLOREMIC METABOLIC ALKALOSIS.
I. INTRODUCTION
Metabolic alkalosis is an acid-base disorder in which the pH of the blood is elevated beyond the normal range of 7.35-7.45. This metabolic condition occurs mainly due to decreased hydrogen ion concentration in the blood, leading to compensatory increased levels of serum bicarbonate, or alternatively, as a direct result of increased bicarbonate concentrations. An elevated PaCO2 is often present as a result of compensatory alveolar hypoventilation.
II. CAUSES OF METABOLIC ALKALOSIS
The five main major causes of metabolic alkalosis are.
III. PHYSIOLOGY OF BICARBONATE HOMEOSTASIS IN THE BODY
Systemic arterial pH is maintained between 7.35 and 7.45 by extracellular and intracellular buffering via respiratory and renal mechanisms [1]. The control of arterial CO2 tension by central nervous system and respiratory system and control of plasma bicarbonate by kidneys stabilize the arterial pH by excretion or retention of acid and alkali. This balance is represented by the Henderson-Hassalbalch equation given by
Where HCO3- represents in the plasma bicarbonate concentration and pCO2 is the plasma carbon dioxide tension in the blood. At normal conditions in the body, the CO2 production and excretion are equal and pCO2 is maintained at 40 mm Hg. At steady state, the bicarbonate exists in different forms within the body which is shown as follows.

Figure 2. Distribution of various forms of bicarbonate in the body. Species of each form are shown as a total percentage of bicarbonate in the body. Primary changes in pCO2 can cause respiratory acidosis or respiratory alkalosis depending on if the value of pCO2 is above or below 40 mm Hg. Primary alteration of pCO2 due respiratory causes cellular buffering and renal adaptation. At the other end, regulation of the metabolic acidotic and alkalotic balance occurs mainly through bicarbonate excretion and resorption in the kidney.
Kidneys regulate plasma HCO3- through 3 main processes [1].
Primary changes in plasma HCO3- due to metabolic or renal factors cause compensatory changes in the ventilation which blunt the changes in the pH. 80-90% of HCO3- produced daily in the body is reabsorbed in the proximal tubule. The distal nephron absorbs the remainder of the HCO3- , thus excreting very small amounts of bicarbonate in the urine daily. However, in cases where this acid-base balance is disturbed, the kidneys possess the capacity to eliminate large amounts of bicarbonate excess in the urine.
IV. PATHOGENESIS OF HYPOCHLOREMIC METABOLIC ALKALOSIS
In the pathogenesis of metabolic alkalosis, the primary abnormality occurs via 2 mechanisms [1,2,3]
The rise in the net reabsorption of bicarbonate through kidney occurs through 3 mechanisms [1, 3].
We will discuss factor 2 mentioned above (chloride depletion and hypochloremia) as relevant to this case.
The gastric acid produced by the gastrointestinal tract contains hydrochloric acid and smaller quantity of potassium chloride. The normal production of hydrochloric acid does not result in metabolic alkalosis in the body since the production of HCl is compensated by the production of bicarbonate from the pancreas once the acidified digested contents enter the duodenum.
Hydrogen loss can occur from the gastrointestinal tract or in the urine. In the presence of vomiting and aspiration of gastric contents, the normal stimulus to the production of the bicarbonate is eliminated which in turn leads to increased levels of bicarbonate in the blood and thus the resulting metabolic alkalosis. The hypochloremia can contribute to the reduction in bicarbonate excretion by increasing distal reabsorption and reducing distal secretion from the kidney. This usually occurs in tandem with the volume depletion which occurs due to the acid loss from the stomach. One milliequivalent of hydrogen ions lost generates one milliequivalent of bicarbonate: the hydrogen ion is derived from water, while the associated hydroxyl ion combines with carbon dioxide to form bicarbonate.
At the cellular level, the balance between the excretion and retention of the bicarbonate in the plasma is maintained by the type A and B cells in the collecting tubules [4, 5]. Bicarbonate reabsorption in the medullary collecting tubule and in type A intercalated cell in the cortical collecting tubule is mediated by hydrogen secretion via H-ATPase pumps and passive cosecretion of chloride in the luminal membrane. Water within the cell dissociates into H+ and OH- ions. H+ ions are secreted into the lumen by H-ATPase pumps in the luminal membrane, where they primarily combine with NH3 to from NH4+. The OH- ions in the cell combine with CO2 to form HCO3- in a reaction catalyzed by carbonic anhydrase. Bicarbonate is then returned to the systemic circulation via Cl-HCO3 exchangers in the basolateral membrane. The favorable inward concentration gradient for Cl- provides the energy for HCO3- reabsorption. The intracellular bicarbonate is returned to the systemic circulation through the Cl/HCO3 exchangers in the basolateral membrane. A decline in the tubular fluid chloride concentration will promote both chloride and hydrogen secretion. H-K-ATPase pumps, which lead to both H+ secretion and K+ reabsorption, are also present in the luminal membrane. The number of these pumps increases with K+ depletion, suggesting that their main function may be to promote K+ conservation.

Fig 3. Electrolyte balance across the type A collecting duct cells in the kidney.
The type B intercalated cells in the cortical collecting tubule are able to directly secrete bicarbonate by reversing the location of the transporters as seen during the recovery phase of metabolic alkalosis [5]. The Cl/HCO3 exchangers are now located in the luminal membrane, leading to bicarbonate secretion into the tubular lumen. The activity of these cells is appropriately enhanced by alkalemia in an attempt to excrete the excess bicarbonate. These two mechanisms play a role in the conservation and excretion of the bicarbonate balance in the body depending on the body pH levels.
V. URINARY ELECTROLYTES IN DIAGNOSIS OF HYPOCHLOREMIC METABOLIC ALKALOSIS
Urine sodium concentration may be used to distinguish between volume depletion (< 25 meq/L) and euvolemia (> 40 meq/L). In metabolic alkalosis volume depletion may not show a low urine sodium concentration [1]. Sodium wasting occurs during the first few days of acute vomiting. Early in the course of vomiting (between 1-3 days), the plasma bicarbonate concentration and therefore the filtered bicarbonate load are increased. The ability to enhance bicarbonate reabsorption takes 3 to 4 days to reach its maximum. Thus, there is increased NaHCO3 delivery to the collecting tubules. Some of the excess sodium is reabsorbed in exchange for potassium in the collecting tubules. This results in the urine which has high urine sodium and potassium concentrations. Potassium losses are the major cause of hypokalemia with vomiting, since the concentration of potassium in gastric secretions is only 5 to 10 meq/L. Also this results in a urine pH above 7.0 due to the bicarbonaturia. The urinary findings change dramatically once NaHCO3 reabsorptive capacity has increased (beyond 4 days), so that all of the filtered bicarbonate can be reabsorbed. The excretion of sodium, potassium, and bicarbonate are all low and the urine pH is paradoxically acidic. A table showing the urine electrolyte changes during the acute and late phases after vomiting is shown below [1].
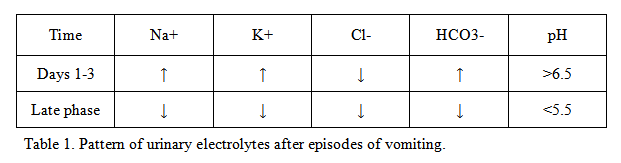
The presence of underlying hypovolemia can be detected by finding a urine chloride concentration below 25 meq/L.Chloride conservation is due to volume depletion, resorption of sodium with chloride and hypochloremia induced by chloride loss in gastric secretions.
VI. RENAL TUBULAR ACIDOSIS AS A CAUSE OF BICARBONATURIA
Elevated bicarbonate levels in the urine may occur in the early phase of metabolic alkalosis (days 1-3) as mentioned above. However, elevated urinary bicarbonate levels may also occur due to renal tubular acidosis which must be remembered in the differential diagnosis. Renal tubular acidosis (RTA) is characterized by the development of metabolic acidosis due to a defect in the ability of the renal tubules to perform normal functions [1, 6]. All forms of RTA are characterized by a normal anion gap (hyperchloremic) metabolic acidosis. This form of metabolic acidosis usually results from either the net retention of hydrogen chloride or its equivalent (such as ammonium chloride) or the net loss of sodium bicarbonate or its equivalent. The major cause of a normal anion gap acidosis in patients without renal failure is diarrhea.
There are three major subgroups of RTA with different clinical characteristics [6]. In type 1 RTA, there is an inability to excrete the daily acid load, resulting, in the absence of alkali therapy, in progressive hydrogen ion retention and plasma bicarbonate concentration that may fall below 10 meq/L. In type 2 RTA, there is bicarbonate wasting only when the plasma bicarbonate concentration is above the bicarbonate reabsorptive threshold. Since the more distal segments have substantial bicarbonate reabsorptive capacity, the plasma bicarbonate concentration is usually between 12 and 20 meq/L in this disorder. In type 4 RTA, the most prominent abnormality is hyperkalemia due to the hypoaldosteronism. The degree of acidosis is generally mild with the plasma bicarbonate concentration being above 17 meq/L. The treatment is dependent on the underlying cause of renal tubular acidosis and the type involved.
VII. SUMMARY AND CONCLUSIONS
In conclusion, this is a case with abnormally elevated levels of bicarbonate in the urine with levels of over 80 meq/L. The case is originally of a hypochloremic metabolic alkalosis due to the presence of a long standing fistula high in the intestinal tract leading to the loss of hydrochloric acid. After admission, the patient was treated with fluid replacement for dehydration. Over 36 hours of the treatment, the patient essentially normalized the alkalotic condition of the body by excreting massive amounts of bicarbonate into the urine which was detected in the laboratory. The patient's fistula was subsequently closed using surgical means resulting in the long term correction of the electrolyte imbalance.
REFERENCES
![]() Contributed by Ramachandra R Gullapalli, MD and Mohamed A Virji, MD, PhD
Contributed by Ramachandra R Gullapalli, MD and Mohamed A Virji, MD, PhD