DIAGNOSIS Embryonal Tumor with Abundant Neuropil and True Rosettes (ETANTR)
DISCUSSION
The 2007 World Health Organization (WHO) classification of central nervous system (CNS) tumors classifies the embryonal tumors into three categories: 1) Medulloblastoma, 2) Atypical teratoid/rhabdoid tumor, and 3) CNS primitive neuroectodermal tumor (PNET). The latter category (PNET) includes five sub-categories: 1) CNS PNET; 2) CNS neuroblastoma, 3) CNS ganglioneuroblastoma, 4) medulloepithelioma, and 5) ependymoblastoma. [7].
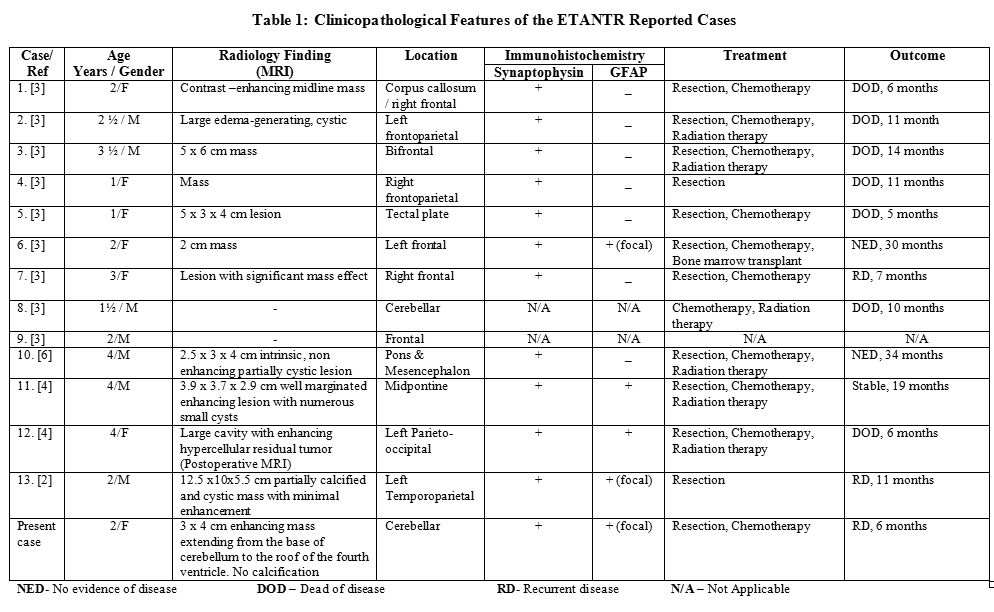
Embryonal tumors with abundant neuropil and true rosettes were first reported as neuroblastic tumors with abundant neuropil and true rosettes in 2000 by Eberhart et al.[3], who described nine cases. In 2006, three additional cases were been reported, one case by Spina et al.[6] and two cases by Fuller et al.[4]. Recently, in 2007, an additional case was reported by Dunham et al.[2]. Table 1 summarizes the features of these reported cases including our case.
In the "CNS PNET" section of the 2007 WHO classification of tumors of the CNS, there is a short paragraph, describing an extremely aggressive type of tumor called "Embryonal tumor with abundant neuropil and true rosettes" (ETANTR), suggesting that this tumor might eventually be considered as a separate entity [7].
All ETANTR cases reported so far have been in children aged 4 years old or less. This tumor combines the features of a neuroblastoma and an ependymoblastoma, by showing fine fibrillary neuropil-like areas admixed with cellular regions and ependymoblastoma-like rosettes. Nevertheless, Homer Wright rosettes, which are the phenotypic hallmark of neuroblastomas, are absent or rarely identified. Eberhart et al.[3] reported nine cases of pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. These cases occurred in children, age 1 to 3 years. The tumor involved the frontoparietal region in six cases, the frontal lobe in one case, the cerebellum in one case and the tectal plate in the last case. Two additional cases were described in an addendum to the article, one involved the cerebellum and the other tumor was in the frontal lobe. Microscopically, their tumors consisted of small to medium-sized, round to oval, hyperchromatic cells with poorly defined cytoplasmic borders. Cells were found in clusters and cords set in a paucicellular fibrillar neuropil matrix. True rosettes, perivascular pseudorosettes and occasional Homer Wright rosettes were present. In our case there were no Homer Wright rosettes identified and there were no perivascular pseudorossettes. Immunohistochemically, their cases were positive for synaptophysin and neurofilament protein and only one case showed focal GFAP positivity [3]. Our case was positive for synaptophysin and CD56 and was focally positive for GFAP but it was negative for neurofilament protein. In their series, the tumors were aggressive and most of the patients died from their disease 5-14 months after initial presentation. There was one patient with recurrent disease 7 months after resection and chemotherapy, which is almost similar to our patient, who developed recurrent disease 6 months after near total resection and chemotherapy. Spina et al.[6] reported a 4-year old boy who presented with a diffuse and infiltrating ETANTR of the pons extending to the midbrain. Histologically, the tumor was similar to that described by Eberhart et al.[3]. Immunohistochemically the tumor cells were positive for synaptophysin and negative for GFAP. Their patient was alive 34 months after surgery with no evidence of disease. Fuller et al.[4] reported two cases that arose in 4-year-old children, one was a midpontine tumor and the other was a large cerebral lesion. The first case showed polysomy of chromosomes 2, 8, 17, and 22. The second case showed isochromosome 17q which is molecular alteration typical of medulloblastomas. The tumor was stable at 19 months of follow-up for the first case while in the second case the patient died of disease 6 months after initial examination. Recently, Dunham et al.[2] reported an additional case occurring in a 2-year-old boy as a large left temporoparietal mass. The tumor exhibited extensive neurocytic differentiation. Their patient developed recurrence approximately after one year. Similar cases have been previously described as ependymoblastoma and neuroblastoma [1, 5, 8].
In summary ETANTR is a very rare, highly aggressive CNS neoplasm and we are reporting here an additional case to the previously reported thirteen cases, which also represents the second case occurring in the cerebellum.
REFERENCES
![]() Contributed by Turki Omar Al-Hussain, MBBS, and Mohammad Anas Dababo, MD
Contributed by Turki Omar Al-Hussain, MBBS, and Mohammad Anas Dababo, MD