FINAL DIAGNOSIS: EHLERS DANLOS SYNDROME TYPE IV (VASCULAR TYPE)
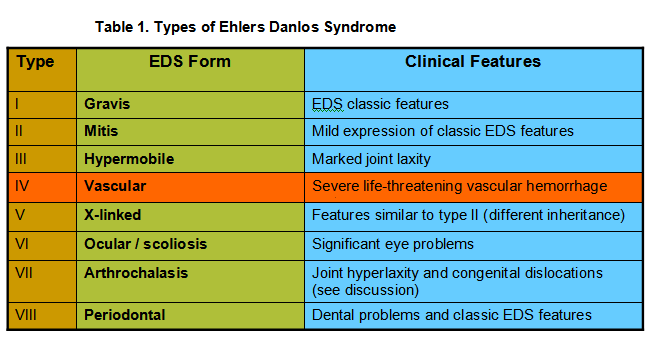
Ehlers Danlos Syndrome (EDS) is a heterogeneous group of heritable connective tissue disorders characterized by joint hypermobility, skin hyperextensibility, cardiac valvular defects and tissue fragility. There are eight major types of EDS (see Table 1), classified according to symptoms and signs. Considering all subtypes, EDS occurs in 1 out of 5000 births.
EDS type IV, named vascular type by Barabas in 1967, is considered a rare autosomal dominant disorder that could present with the typical signs of other EDS types along with easy bruising and hematomas in areas of trauma, and translucent skin (visible veins). Hypermobility of large joints and hyperextensibility of the skin, characteristic of other forms of EDS, are unusual in the vascular type. For all EDS subtypes, type IV accounts for less than 4% of these disorders, but is clearly the most clinically severe type. The precise incidence and prevalence is not known. The diagnosis is often difficult and mainly based on clinical criteria and family history. Some consider that the diagnosis can be achieved by having two of the following major criteria: thin, translucent skin; arterial, intestinal or uterine fragility or rupture; extensive bruising; characteristic facial appearance. According to some studies, skin hyperextensibility is not a major feature of EDS type IV. These patients are at risk of gastrointestinal, uterine, and arterial rupture. Most common causes of death are rupture of abdominal aorta, colon perforation (as in our patient) and cerebral bleeding. Many reports emphasize the exceedingly high risk of massive bleeding and anastomotic disruption with attempted operative repair. Joint hypermobility is determined by (1) passive apposition of thumbs to the flexor aspect of the forearm, (2) passive hyperextension of the fingers parallel to the extensor aspect of the forearms, (3) active hyperextension of the elbows >10ˇă beyond 180ˇă; (4) active hyperextension of the knees >10ˇă beyond 180ˇă; (5) ability to place palms on the floor with knees extended. A scoring system named the Beighton scale helps to determine the degree of hypermobility.
A few reports have described some unusual phenotypic manifestations, including large eyes with a thin nose, hollow cheeks, small lips, lobeless ears, short stature, and thin scalp hair. Other skin manifestations are elastosis serpiginosa (scar formation after trauma with serpiginous keloid formation), keloid formation, Raynaud phenomenon, and acrogeria that refer to excessive wrinkling and thinness of the skin over hands and feet, which may produce an elderly appearance. Vascular and visceral complications develop in 25% cases before 20 years of age and 80% cases before age 40.
Oral manifestations (gingival contraction) have been reported, as well although these are more characteristic of EDS types VII and VIII. Type IV of EDS has variable penetrance and expressivity of the disease, and some of the clinical features may not be present. This variability is probably related to the amount of type III collagen synthesized by the patients. Oderich et al reported the median age of EDS type IV disease diagnosis was 28.5+/-11 years in 31 patients evaluated during the period of 1971 - 2001. In the absence of vascular complications, survival was 90% at 20 years but 20% at 60 years. Three of these patients died from exsanguination hemorrhage, two in the operating room and one due to rupture of the splenic artery.
Vascular complications affected medium and large-sized arteries in many anatomic locations in EDS type IV. Most lesions are located in the aorta and its proximal branches or within the visceral and renal arteries. The exact nature of the vascular lesions has been a matter of debate. Most represent arterial dissections, with associated aneurysmal degeneration or arterial tears in contained hematomas, false aneurysms or intracavitary bleeding. Ninety-two percent (92%) of deaths in EDS type IV are due to vascular complications. In another study, Pepin and colleagues stated that in 419 patients with EDS type IV (220 patients and 199 relatives), 272 had arterial complications. No effective intervention is available to decrease the risk of vascular complications. Operative treatment is indicated if there is life threatening bleeding.
Pepin and colleagues also observed that most of the identified bowel complications (62 of 87 patients) occur in the colon (predominantly sigmoid colon, 29 individuals). Perforation of the small bowel (7 subjects) and stomach (2 subjects) was uncommon. Ten deaths occurred due to rupture of the gastrointestinal tract. Tissue fragility and poor wound healing contributed to surgical complications, death or both. Recurrent bowel rupture (15 individuals) occurred in between 2 weeks and 26 years after the first event. Spontaneous bowel perforation was treated by partial colectomy, in 42 subjects, 7 had a second bowel perforation. In this same study, from a total of 183 cases with pregnancy, 3 stillbirths and 3 voluntary terminations were observed in 167 surviving term infants. Twelve women died during the peripartum period from complications of their EDS type IV disease.
Our patients (CASE 1 and CASE 2) presented a clinical picture highly suggestive of ED type IV disease. The history of abdominal pain, frequent nose bleeds, easy bruising and recurrent hematoma was typical of EDS type IV in case 1. His surgical complications and friable tissue status supported the diagnosis. Similarly, abdominal pain and recurrent hematoma in case 2, supported by a positive family history of sudden death associated with abdominal aneurysm, was strongly suspicious for type IV EDS. Aneurysms and recurrent unexplained bruises appear pathognomonic for this subtype of EDS, and was confirmed in our 2 cases (in both cases by molecular analysis, case 2 also by biochemical collagen analysis).
Etiology
EDS type IV is associated with heterozygous mutations in the COL3A1 gene located on the long arm of Chromosome 2, which encodes type III pro-collagen, a part of vessels and organs structure. The distribution of type III collagen is predominantly in skin, walls of blood vessels, and hollow organs. Normal type III collagen is a fibrillar protein composed of 3 ¦Á1 (III) polypeptide chains. It is a major component of the extracellular matrix in many organs and the skin and is essential for normal collagen I fibrillogenesis in the cardiovascular and other organ systems. COL3A1 mutations produce a mutant pro ¦Á1 (III) polypeptide that lead to in an abnormal type III collagen. EDS type IV may result then from a variety of mutations in the COL3A1 gene, including point mutations, deletions, insertions and splicing mutations. At present, there is no phenotype / genotype correlation.
Collagen has an unusual amino acids composition: Glycine (Gly) is found at almost every third residue and proline (Pro) constitutes about 9% of collagen amino acid composition. Hydroxyproline (Hyp) is derived from proline. Hydroxylysine is derived from lysine. Most collagen forms in a similar manner, but the following process is typical for collagen type I (see figure 1). Inside the cell three peptide chains are formed (2 alpha-1 and 1 alpha-2 chain) in ribosomes along the Rough Endoplasmic Reticulum (RER). These peptide chains (or preprocollagen), that contain 'signal' and 'registration' peptides, migrate into the lumen of the RER. Signal Peptides are cleaved inside the RER and the chains are referred as procollagen. Hydroxylation of lysine and proline amino acids occurs within the lumen. This process is dependent open Vitamin C (ascorbic acid) as a cofactor. Glycosylation of hydroxylated amino acid occurs. Inside the RER a triple helical structure is formed. Procollagen travels to the golgi apparatus, where it is packaged and secreted by exocytosis. Outside the cell registration peptides are cleaved and tropocollagen is formed. Multiple tropocollagen molecules form collagen fibrils, and these further coalesce to form collagen fibers. Collagen then attaches to cell membranes via other proteins, such as fibronectin and integrin.

Laboratory
Most reports agree that the clinical diagnosis is strongly suggestive, but laboratory confirmation is advisable. Histology and electron microscopy are not diagnostic (arterial wall thinning, decrease collagen content, distorted collagen architecture). The most reliable study is direct assessment of procollagen III by protein gel electrophoresis. The laboratory diagnosis relies on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) with fibroblasts for the detection of type III collagen. The method requires a skin biopsy, but is more sensitive and less costly than the COL3A1 gene analysis. The nature of the COL3A1 gene mutation does not accurately predict the extent and prognosis of EDS type IV.
If imaging studies are performed to rule out the presence of an arterial dissection or rupture, arteriography should be avoided and non-invasive procedures such as ultrasound and CT scans are preferred. For the molecular studies, RNA and DNA are extracted from cultured fibroblasts and complementary DNA is synthesized by reverse transcription from RNA. Overlapping fragments of complementary DNA are amplified by PCR (polymerase chain reaction) and analyzed by electrophoresis on agarose gels to identify gene abnormalities. Abnormal fragments detected in this way can be re-amplified from genomic DNA and sequenced by the dideoxy chain-termination method with a T4 polymerase.
All mutations should be confirmed by sequencing analysis or restriction enzyme digestion of genomic DNA. Pepin et al, in 135 patients, identified the causative mutations in the COL3A1 gene. Four mutations resulted in deletion of multiple exons, 41 of them resulted in skipping of a single exon. In seven patients, one mutation (IVS24 + 1G ' A) led to skipping of exon 24. Four patients had splice-site mutations with complex splicing outcomes and multiple messenger RNA occured. A 10-bp deletion in the acceptor site of intron 29 in one patient resulted in a cryptic site within exon 30 and deletion of three amino acids from the triple helix. In the remaining 85 patients, 73 different point mutations led to substitution of various amino acids for conserved glycine residues throughout the triple-helical domain. In 7 families, G16S mutation was identified. G82D, G373R, G385E, G415S, G499D and G1021E was detected in two families.
The precise COL3A1 gene mutation described in CASE 2 (IVS27 + 1 G'C) has not been reported before. Based on what is known about exon splicing and the understanding of the consequences of another intron 27 splice donor site mutation, this mutation is predicted to result in skipping of exon 27 in transcripts associated with the mutant allele. Therefor, this mutation probably alters a consensus splice donor GC ' CG, meaning a shipping of exon 27 in the splice polyA+RNA and deletion of 18 amino acids.
Genetic Counseling
The implication of a dominant pattern of inheritance implies that affected individuals have a 50% chance of transmitting the mutated gene to each child. Therefore, genetic counseling is warranted. Families should be provided with information of contraception, reproductive and pregnancy risks, management of inadvertent pregnancies and options of adoption and insemination with donor sperm.
REFERENCES:
![]() Contributed by Rosemary Recavarren, MD, Mohamed Virji, MD, PhD, Lorna Cropcho and K. Michael Gibson, MD
Contributed by Rosemary Recavarren, MD, Mohamed Virji, MD, PhD, Lorna Cropcho and K. Michael Gibson, MD