![]() Contributed by Sara E Monaco, MD and Jeffrey Kant, MD, PhD
Contributed by Sara E Monaco, MD and Jeffrey Kant, MD, PhD
![]() Published on line in December 2006
Published on line in December 2006
PATIENT HISTORY:
This 3-month-old girl was referred to the Neurology Clinic for hypotonia. The child was born at 36 weeks to a 22-year-old G1P0 mother with an uncomplicated pregnancy. The neonatal period was uneventful, however, in the first few months, the pediatrician noted that the child seemed hypotonic.
The physical examination at 3 months revealed a height and weight in the 10-25th percentile. The child appeared well-nourished and was noted to have an unusual breathing pattern. On review of systems, the patient reportedly had reached certain developmental milestones, such as the ability to smile, coo, follow, and grasp. However, she was not able to lift her head, roll over, or lift her legs. She was only able to move her arms weakly. The mother did not notice any problems in the child's ability to swallow or gain weight, and there were no reported seizures or bowel/bladder dysfunction.
A neurological examination demonstrated weak strength overall with pronounced difficulty lifting her arms and no ability to lift her legs. There was also a decrease in passive and active tone, and she was areflexic. In addition, tongue fasciculations were noted. Sensation and cranial nerves were intact. Family history was unremarkable.
PATIENT WORK-UP:
EMG: Positive evidence for denervation
Genetic Testing: PCR amplification for homozygous deletion of SMN genes (autosomal recessive spinal muscular atrophy; SMA type 1; Werdnig Hoffman Disease).
THEORETICAL BASIS OF GENETIC TEST FOR SMA:
A portion of the SMN, or "survival motor neuron" locus, is absent in the majority of SMA patients. This gene, located on the long arm of chromosome 5, consists of 9 exons. The SMN gene exists as 2 highly homologous copies in an inverted, duplicated region on the long arm of chromosome 5: SMN1 (telomeric copy) and SMN2 (centromeric copy). The 2 copies of SMN differ by 5 nucleotide base pairs (one in exon 7, exon 8, and intron 6; two in intron 7). The single base pair differences in exon 7 and 8 can be used to distinguish SMN1 and SMN2 in a PCR-RFLP assay (see discussion below). Since SMN1 and SMN2 can each be detected by this method, it is possible to detect the homozygous absence of SMN1 seen in most SMA patients.
ASSAY PRINCIPLE AND INTERPRETATION:
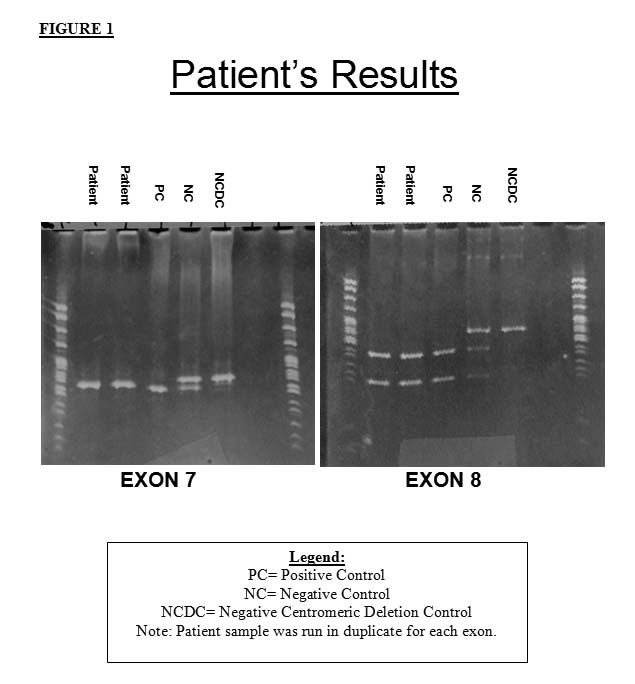
Genomic DNA isolated from the sample provided (eg blood, tissue, other) undergoes PCR amplification of exons 7 and 8 of the centromeric and telomeric SMN gene copies. PCR products from each exon are individually subjected to digestion by an appropriate restriction enzyme, fragments separated on an acrylamide gel and visualized following staining with a DNA-binding dye.
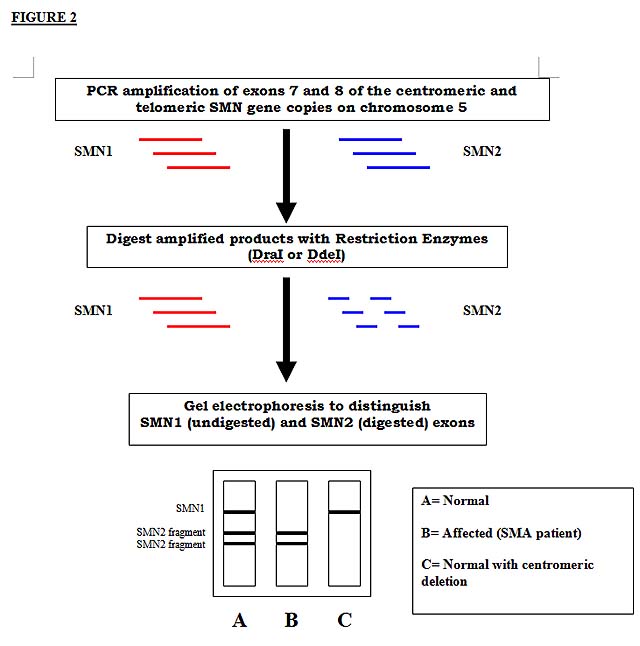
The PCR products from exon 8 of the 2 SMN gene copies can readily be distinguished since the centromeric (SMN2) gene contains a recognition site for the restriction enzyme DdeI that is absent in exon 8 of the telomeric SMN. After restriction enzyme digestion, the SMN1 exon 8 product will be undigested (high molecular weight, travel less distance in the gel) and the SMN2 exon 8 product will be digested into 2 different sized fragments of about 122 and 78 base pairs in length.

In exon 7, the situation differs slightly. Since there is no native restriction enzyme recognition site in exon 7, a mismatched oligonucleotide primer can be used to specifically introduce a nucleotide difference. This nucleotide change introduced by the primer, in combination with the native single nucleotide difference, creates a DraI site in the SMN2 PCR product. Thus, after digestion with the restriction enzyme, the SMN1 exon 7 product will be undigested, whereas the SMN2 exon 7 product will be digested into 2 fragments of 176 and 24 base pairs. The 24 base pair fragment migrates off the gel leaving a single undigested band of SMN1 exon 7 and the larger digested fragment of SMN2 exon 7.

The patient's sample revealed the expected results for a patient with SMA. The exon 7 gel shows a single digested fragment of the SMN2 gene, but no SMN1 gene product. The exon 8 gel shows two fragments corresponding to the digested SMN2 gene, but no SMN1 gene product. Thus, there appears to be homozygous absence of the SMN1 gene by PCR-RFLP analysis.
ASSAY LIMITATIONS: