Final Diagnosis -- A 7 year old boy with microcytosis with pancytopenia
FINAL DIAGNOSIS:
Peripheral Blood
Microcytosis with pancytopenia.
Bone Marrow Aspirates and Biopsy
Normocellular marrow with trilineage hematopoiesis and atypical macrophages.
Iron stores present.
The diagnostic comment includes:
The bone marrow demonstrates the presence of numerous macrophages with Gaucher-type features; these cells demonstrate voluminous cytoplasm with a crumpled tissue paper type appearance. The findings could be compatible with marrow involvement by Gaucher disease, but would require correlation with results of pending enzyme testing for confirmation of this diagnosis.
FOLLOW UP:
An enzyme panel demonstrated normal activities for all lysosomal enzymes with the exception of beta glucosidase (glucocerebrosidase), which was low. These findings were interpreted as presumptive Gaucher disease, but confirmation was recommended with cultured fibroblasts because protein levels were low in the corresponding leukocyte extract. Molecular studies are proposed to determine the patient's mutation.
Gaucher disease was first described by Philippe Gaucher in 1882, who hypothesized that infiltration of enlarged cells in a spleen represented a neoplasm.1 Gaucher disease is the most prevalent lysosomal storage disorder and is inherited in an autosomal recessive fashion.2 It is most common in the Ashkenazi Jewish population. In a population survey involving 2000 Ashkenazi Jews, the gene frequency was 0.034.3 Approximately 6.8% of the Jewish population is heterozygous for Gaucher disease, and, based on this, the expected birth frequency is 1 in 1000.1 The disease is also relatively common among the Norbottnian population in Northern Sweden.
DISCUSSION:
 Gaucher disease was first described by Philippe Gaucher in 1882, who hypothesized that infiltration of enlarged cells in a spleen represented a neoplasm.1 Gaucher disease is the most prevalent lysosomal storage disorder and is inherited in an autosomal recessive fashion.2 It is most common in the Ashkenazi Jewish population. In a population survey involving 2000 Ashkenazi Jews, the gene frequency was 0.034.3 Approximately 6.8% of the Jewish population is heterozygous for Gaucher disease, and, based on this, the expected birth frequency is 1 in 1000.1 The disease is also relatively common among the Norbottnian population in Northern Sweden.
Gaucher disease was first described by Philippe Gaucher in 1882, who hypothesized that infiltration of enlarged cells in a spleen represented a neoplasm.1 Gaucher disease is the most prevalent lysosomal storage disorder and is inherited in an autosomal recessive fashion.2 It is most common in the Ashkenazi Jewish population. In a population survey involving 2000 Ashkenazi Jews, the gene frequency was 0.034.3 Approximately 6.8% of the Jewish population is heterozygous for Gaucher disease, and, based on this, the expected birth frequency is 1 in 1000.1 The disease is also relatively common among the Norbottnian population in Northern Sweden.
Differential Diagnosis
Pseudo-Gaucher cells, which are found in many hematologic abnormalities, including CML, AML, CLL, Hodgkin lymphoma, multiple myeloma, and ITP, cannot be distinguished from Gaucher cells on H&E stained sections. However, Gaucher cells show diffuse iron staining, while pseudo-Gaucher histiocytes are generally negative.4 In pseudo-Gaucher cells, cellular debris accumulates when the capacity of lysosomes is overwhelmed by the material produced from apoptosing cells.5
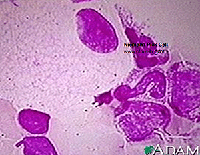 Niemann-Pick disease is caused by a deficiency in sphingomyelinase which leads to accumulation of sphingomyelin in the cytoplasm of macrophages. The globules are small and relatively uniform in size, sometimes described as mulberry-like in appearance. Foamy macrophages similar to Niemann-Pick cells may be seen in any disorder that is associated with massive cell destruction that overloads the capacity of the cellular machinery to digest lipids (e.g., thalassemias, sickle cell anemia, ITP, chronic renal failure).5 The vacuoles may be larger and more irregular in size than those seen in true Niemann-Pick cells. As compared to Gaucher cells, the cytoplasm in these macrophages is foamy and vacuolated as opposed to fibrillary.
Niemann-Pick disease is caused by a deficiency in sphingomyelinase which leads to accumulation of sphingomyelin in the cytoplasm of macrophages. The globules are small and relatively uniform in size, sometimes described as mulberry-like in appearance. Foamy macrophages similar to Niemann-Pick cells may be seen in any disorder that is associated with massive cell destruction that overloads the capacity of the cellular machinery to digest lipids (e.g., thalassemias, sickle cell anemia, ITP, chronic renal failure).5 The vacuoles may be larger and more irregular in size than those seen in true Niemann-Pick cells. As compared to Gaucher cells, the cytoplasm in these macrophages is foamy and vacuolated as opposed to fibrillary.
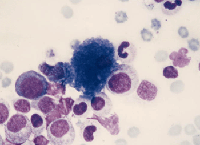 Sea blue histiocytes may be found in small numbers in normal bone marrow. They are increased in any disorder associated with massively increased intramedullary cell destruction, such as chronic myelogenous leukemia. The macrophage cytoplasm is filled with insoluble lipid pigment, called ceroid, which is thought to represent partially digested globosides derived from cell membranes.5 As compared to Gaucher cells, these cells stain more intensely blue with Wright-Giemsa and the inclusions are globular rather than fibrillary.
Sea blue histiocytes may be found in small numbers in normal bone marrow. They are increased in any disorder associated with massively increased intramedullary cell destruction, such as chronic myelogenous leukemia. The macrophage cytoplasm is filled with insoluble lipid pigment, called ceroid, which is thought to represent partially digested globosides derived from cell membranes.5 As compared to Gaucher cells, these cells stain more intensely blue with Wright-Giemsa and the inclusions are globular rather than fibrillary.
Pathogenesis of Gaucher Disease
Enzymatic degradation of cell constituents takes place primarily in secondary lysosomes. Secondary lysosomes, also known as phagolysosomes, form when primary lysosomes fuse with phagocytic vacuoles containing the ingested material.1 Gaucher disease is due to a hereditary deficiency in the activity of glucocerebrosidase, one of the lysosomal enzymes required for glycolipid degradation.
Macrophages are the primary cells affected, and include all cells of the mononuclear phagocyte system, especially Kupffer cells in the liver, osteoclasts in bone, microglia and CSF macrophages in the central nervous system, alveolar macrophages in the lungs, and histiocytes in the spleen, lymph nodes, bone marrow, gastrointestinal and genitourinary tracts, as well as the peritoneum. They have been described in almost every organ in the body.6 However, the mechanism by which glucocerebroside accumulation leads to the cellular pathology in Gaucher disease remains undefined.
An illustration demonstrating how cytoplasmic material accumulates in Gaucher and Niemann-Pick diseases can be seen in ""Color Atlas of Hematology" 1998: 327)
Genetics
The glucocerebrosidase gene is located on chromosome 1q21 and over 200 mutations associated with Gaucher disease have been identified.7 In the Ashkenazi Jewish population, the predominant mutation is 1226A --> G, resulting in an Asp --> Ser substitution at amino acid 370 (N370S). This mutation accounts for ~75% of the mutant alleles in Jewish patients and ~30% in non-Jewish patients.1 It predisposes to type 1 disease and precludes neurological involvement. Compound heterozygosity with N370S predicts a more severe disease, although there is still absence of neurological symptoms.8 A frameshift mutation resulting in the insertion of a guanine at nucleotide 84 (84GG) is also common in the Jewish population. The 1448T?C (L444P) mutation is common in the Norbottnian population and the homozygous state has a very high association with the neuronopathic (see below) variants of Gaucher disease.8 The five most common mutations account for ~97% of the alleles in the Jewish population, but ~75% in the non-Jewish.1
Clinical Features
There are three major types of Gaucher disease, distinguished primarily by the presence or absence of neuronopathic manifestations. Type 1 (Adult, or Non-neuronopathic) accounts for 99% of cases. Although referred to as "adult," it may occur at any age and, as its name implies, is characterized by absence of neurologic symptoms. Manifestations range from asymptomatic hepatosplenomegaly to the development of anemia, thrombocytopenia, neutropenia, and bony involvement.6 Type 2 (Acute infantile neuronopathic) shows onset of neurologic effects within the first six months with rapid neurologic deterioration and early death, usually by the age of two years.2 In type 3 (Juvenile or Subacute neuronopathic), onset of neurologic disease may occur within the first three months, but there is delayed or little progression for several years or decades. Patients affected by this type may live into their forties. The prototype is the Norbottnian form.6
Gaucher disease can affect most organ systems, and its manifestations are highly variable.7 Mild to massive splenomegaly is usually present and may lead to findings associated with hypersplenism. Anemia and thrombocytopenia are found in almost all cases, while leucopenia is variable. In the absence of hypersplenism, pancytopenia is thought to result from bone marrow infiltration with Gaucher cells.9 Although anemia in Gaucher patients is usually due to red cell destruction, iron deficiency is not an uncommon finding, particularly among younger patients. Our patient's iron indices, except for the normal ferritin level, were consistent with iron deficiency. Ferritin is often increased in Gaucher disease, and it may have been in the normal range in our patient due to Gaucher-associated elevation.9 Therefore, our patient's anemia may be due, in part, to iron deficiency.
The liver is proportionately less enlarged than the spleen in patients with Gaucher disease, and liver function tests are usually normal.9 However, there still may be mechanical symptoms due to enlargement. Also, liver fibrosis may develop and is sometimes severe enough to cause esophageal varices. A variety of pulmonary findings may also be present. In one autopsy series, manifestations of lung involvement were found in three patterns: 1) interstitial infiltration of Gaucher cells with fibrosis; 2) filling of alveolar spaces with Gaucher cells and alveolar consolidation; and 3) capillary plugging by Gaucher cells with secondary pulmonary hypertension.6
In the skeletal system, patchy areas of bone demineralization and areas of infarction develop in greater than 20% of cases. Bone lesions with accompanying pain may be the most debilitating feature of type 1 disease. Widening of the distal femur may give rise to an "Erlenmeyer flask" deformity.1 Aseptic necrosis of the femoral heads and vertebral collapse are also common.
Neurologic manifestations are the hallmark of types 2 and 3, and may include oculomotor abnormalities, hypertonia of the neck muscles with opisthotonus, bulbar signs, limb rigidity, seizures, and sometimes choreoathetoid movements. Type 3 disease is further subdivided into a, b, and c based upon neurologic features.6
Increased risk of neoplasm has also been considered. A retrospective study involving 48 Gaucher patients and 511 controls, revealed a 14.7 fold increase in risk of cancers of hematopoietic origin.10 This study has been criticized for possible ascertainment bias. Another retrospective study, with 476 patients, found an increased incidence of multiple myeloma up to 5.5 fold, but only a slight increase in other hematological malignancies.9 Therefore, there may be an increased risk of some types of hematological malignancies, but additional investigation is required to further elucidate the true extent of this risk.
Diagnosis
Diagnosis was classically based on identification of Gaucher cells in a bone marrow aspirate. However, enzymatic assay to evaluate beta-glucosidase (glucocerebrosidase) activity in leukocytes or cultured fibroblasts combined with molecular analysis is now the gold standard.2,7 Type 1 demonstrates reduced but detectable levels of glucocerebrosidase activity while type 2 shows virtually no detectable activity. Chitotriosidase, a human analog of chitinase produced by activated macrophages, has been used to monitor Gaucher disease severity and improvement upon treatment.2 Chitotriosidase is produced specifically by storage cells, particularly Gaucher cells, but not common tissue macrophages or dendritic cells. Plasma chitotriosidase reflects the total level of enzyme secreted by Gaucher cells throughout the body, and therefore correlates with the number and activity of these cells.11
Treatment
Enzyme replacement therapy with a recombinant product known as imiglucerase (Cerezyme) is the mainstay of treatment. Hepatosplenomegaly, anemia, and thrombocytopenia usually improve within 6 months.1 Unfortunately, there is no effect on neurological symptoms because it does not cross the blood-brain barrier.12 Substrate reduction therapy with inhibitors of glucocerebroside formation has been approved for patients in whom enzyme replacement therapy is "unsuitable" or "not a therapeutic option." 2 Miglustat (N-butyldeoxynojirimycin) is the only substrate reduction intervention thus far licensed, and causes a reduction in hepatosplenomegaly, but a slower response of hematological parameters. Future treatments may include gene therapy and intervention with chemical chaperones. The latter have demonstrated some capacity to stabilize or reactivate improperly folded glucocerebrosidase in vitro.1
The following table summarizes much of the information covered in this case:
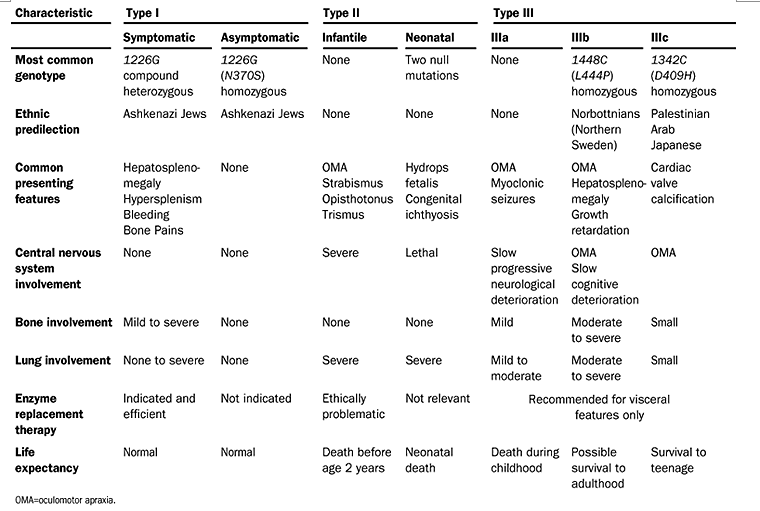
Elstein et al. Lancet 2001;358:324-327.
REFERENCES
- Beutler E. Gaucher disease. In: Williams Hematology. Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT, eds. New York, NY: McGraw-Hill 2006.
- Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. BJH 2005;129:178-188.
- Beutler E, Nguyen NJ, Henneberger MW, et al. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet 1993;52:85-88.
- Weisberger J, Emmons F, Gorczyca W. Images in haemotology. BJH 2004;124:696.
- Cytoplasmic inclusions in bone marrow cells. In: Color Atlas of Hematology. Glassy E, ed. Northfield, IL: CAP 1998:320-327.
- Beutler E, Grabowski GA. Gaucher disease. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, eds. New York, NY: McGraw-Hill 2001:3635-3668.
- Elstein D, Abrahamov A, Hadas-Halpern I, Zimran A. Gaucher's disease. The Lancet 2001;358:324-327.
- Grabowski G. Recent clinical progress in Gaucher disease. Curr Op Ped 2005;17:519-524.
- Zimran A, Altarescu G, Rudensky B, Abrahamov A, Elstein D. Survey of hematological aspects of Gaucher disease. Hematology 2005;10:151-156.
- Shiran A, Brenner B, Laor A, Tatarsky I. Increased risk of cancer in patients with Gaucher disease. Cancer 1993;72:219-224.
- Aerts JMFG, Hollak CEM, van Breeman M, Maas M, Groener JEM, Boot RG. Identification and use of biomarkers in Gaucher disease and other lysosomal storage diseases. Acta Paediatrica 2005;94:43-46.
- Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med 2006;57:283-296.
 Contributed by Siobhan O’Connor, MD, Lydia Contis, MD, K Michael Gibson, PhD, David Finegold, MD and Mohamed Virji, MD, PhD
Contributed by Siobhan O’Connor, MD, Lydia Contis, MD, K Michael Gibson, PhD, David Finegold, MD and Mohamed Virji, MD, PhD



