Final Diagnosis -- Anemia
FINAL DIAGNOSIS:
Peripheral Blood
Macrocytic anemia with bizarre nucleated red blood cells.
Severe thrombocytopenia.
Bone Marrow
Hypercellular bone marrow with megaloblastic anemia.
Reduced trilineage hematopoiesis with marked erythroid predominance.
Stainable iron is present.
DISCUSSION:
Macrocytic anemia may be classified as megaloblastic, due to impaired DNA synthesis in erythrocyte precursors, or nonmegaloblastic, due to various other etiologies (Table 1) [1].
Table 1.
|
Megaloblastic Causes |
Nonmegaloblastic
Causes |
|
Vitamin B12 and/or
folate deficiency
Bone marrow
disorders
Drug-induced
disorders of DNA synthesis
Anticonvulsant
agents (phenytoin, primidone,
phenobarbital)
Oral
contraceptives
Chemotherapeutic
agents (cyclophosphamide,
methotrexate,
hydroxyurea)
Pyrimethamine
Triamterene
Sulfasalazine
Sulfamethoxazole
Trimethoprim
Zidovudine |
Alcoholism (without
folate deficiency)
Accelerated
erythropoiesis
Hemolytic anemia
Posthemorrhagic
anemia
Increased red blood
cell membrane surface area
Obstructive
jaundice
Hepatic disease
Postsplenectomy
Bone marrow
disorders
Chronic obstructive
pulmonary disease
Hypothyroidism |
 In the Western world, about 90% of megaloblastic anemias result from either alcohol-induced folate deficiency or B12 deficiency [1]. Vitamin B12 and folic acid are coenzymes required for synthesis of thymidine. Impaired metabolism or deficiency of these vitamins leads to defective nuclear maturation due to abnormal or inadequate DNA synthesis, with an associated delay or block in cell division [2].
In the Western world, about 90% of megaloblastic anemias result from either alcohol-induced folate deficiency or B12 deficiency [1]. Vitamin B12 and folic acid are coenzymes required for synthesis of thymidine. Impaired metabolism or deficiency of these vitamins leads to defective nuclear maturation due to abnormal or inadequate DNA synthesis, with an associated delay or block in cell division [2].
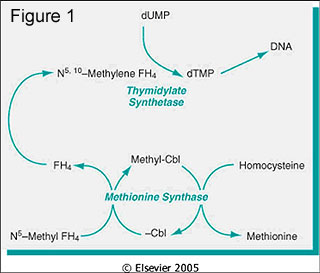
Folic acid is derived from green vegetables and certain fruits. It (or more correctly, tetrahydrofolate [FH4] derivatives) serves to transfer one-carbon units to various compounds, and acts as an acceptor of one-carbon fragments from compounds. Important metabolic processes are dependent on these one-carbon transfers, including 1) purine synthesis; 2) conversion of homocysteine to methionine (also requires vitamin B12); and 3) deoxythymidylate monophosphate synthesis. Deoxythymidylate monophosphate is required for DNA synthesis (Figure 1), and folate deficiency, thus, results in impaired DNA synthesis [2].
Our patient had normal serum and RBC folate levels, and the presumed etiology of the anemia is his profoundly low vitamin B12 level. Vitamin B12 is derived from foods of animal origin. Methylcobalamin is an essential cofactor for methionine synthase, an enzyme involved in the conversion of homocysteine to methionine (as shown in Figure 1). In the process, N5-methyl FH4 is converted to tetrahydrofolic acid (FH4) [2]. As described above, FH4 is essential in DNA synthesis. It is believed that the fundamental cause of defective DNA synthesis in vitamin B12 deficiency is the reduced availability of FH4, which is "trapped" as N5-methyl FH4 [2].
While vitamin B12 deficiency has a number of causes (Table 2), the most common is pernicious anemia [3].
Table 2.
|
Vitamin B12 Deficiency |
|
Decreased intake
Inadequate diet, vegetarianism
Impaired absorption
Intrinsic factor deficiency
Pernicious anemia
Gastrectomy
Malabsorption states
Diffuse intestinal disease, e.g., lymphoma, systemic
sclerosis
Ileal resection, ileitis
Competitive parasitic uptake
Fish tapeworm infestation
Bacterial overgrowth in blind loops and diverticula of
bowel
Increased requirement
Pregnancy,
hyperthyroidism, disseminated cancer |
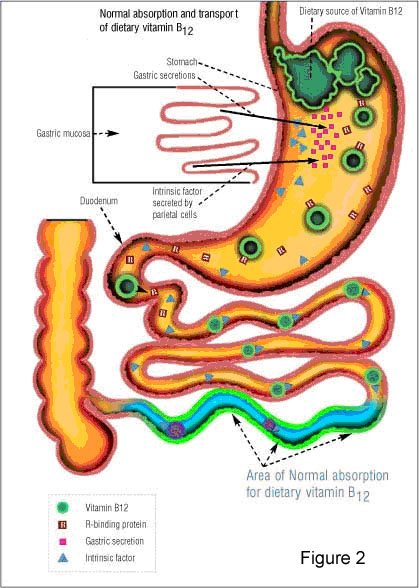 The term "pernicious anemia" refers specifically to the condition associated with chronic atrophic gastritis. Thomas Addison first described pernicious anemia in 1849, and in 1860, Austin Flint linked the anemia to the stomach. Cooked liver successfully treated the anemia, suggesting that it was caused by an extrinsic factor in liver combined with an intrinsic factor found in gastric juice [3]. Type A gastritis, which involves the fundus and body of the stomach, sparing the antrum, is associated with pernicious anemia, autoantibodies to gastric parietal cells and intrinsic factor, achlorhydria, low serum pepsinogen I levels, and high serum gastrin concentrations [3]. Intrinsic factor is a 60-kd glycoprotein produced by gastric parietal cells that binds vitamin B12. The vitamin B12-intrinsic factor complex is absorbed in the terminal ileum, after binding to intrinsic-factor receptors on the luminal membranes of ileal cells (Figure 2) [3].
The term "pernicious anemia" refers specifically to the condition associated with chronic atrophic gastritis. Thomas Addison first described pernicious anemia in 1849, and in 1860, Austin Flint linked the anemia to the stomach. Cooked liver successfully treated the anemia, suggesting that it was caused by an extrinsic factor in liver combined with an intrinsic factor found in gastric juice [3]. Type A gastritis, which involves the fundus and body of the stomach, sparing the antrum, is associated with pernicious anemia, autoantibodies to gastric parietal cells and intrinsic factor, achlorhydria, low serum pepsinogen I levels, and high serum gastrin concentrations [3]. Intrinsic factor is a 60-kd glycoprotein produced by gastric parietal cells that binds vitamin B12. The vitamin B12-intrinsic factor complex is absorbed in the terminal ileum, after binding to intrinsic-factor receptors on the luminal membranes of ileal cells (Figure 2) [3].
In patients with pernicious anemia, malabsorption of vitamin B12 is caused by intrinsic factor deficiency. This is due to two mechanisms: 1) the progressive destruction of parietal cells from the gastric mucosa leading to intrinsic factor destruction and 2) the binding of blocking autoantibodies to the vitamin B12-binding site of intrinsic factor, preventing the formation of the vitamin B12-intrinsic factor complex [3]. If confirmation of malabsorption due to intrinsic factor deficiency is needed, the Schilling test can be performed. However, parietal cell antibodies and intrinsic factor antibodies are now being used to diagnose autoimmune gastritis and pernicious anemia, respectively [4].
In our patient, the source of vitamin B12 deficiency is presumed to be the ileal resection in 1999. Any condition associated with ileal dysfunction may lead to vitamin B12 malabsorption and deficiency [5]. Diseases include celiac disease, tropical sprue and Crohn's disease. In Crohn's disease, the absorption is dependent on the extent of the mucosal lesion and whether the patient has undergone ileal resection. Vitamin B12 malabsorption almost always occurs if the resection involves greater than 100 cm. The body requires about 1 g of B12 daily. The typical diet provides 5 to 15 g daily, and the liver stores a maximum of 2,000 to 5,000 g. Vitamin B12 deficiency therefore develops many years after B12 absorption ceases. If a patient requires lifelong administration of B12, discontinuation will result in deficiency in about 5 years [1]. Therefore, the six year interval since our patient's ileal resection is consistent with the amount of time needed to deplete vitamin B12 reserves.
It is important for several reasons to establish whether vitamin B12 deficiency is the etiology of a megaloblastic anemia, and to distinguish it from folate deficiency. Vitamin B12 deficiency is easily treated with subcutaneous, or, more recently, oral cyanocobalamin (vitamin B12). Even more importantly, vitamin B12 deficiency may lead to peripheral neuropathy and lesions in the posterior and lateral columns of the spinal cord, and in the cerebrum. The most frequent manifestations are paresthesias and numbness [3]. Therapy with folic acid can mask the B12 deficiency and lead to irreversible neurological damage [4]. Diagnosis of B12 deficiency can usually be made if low serum vitamin B12 is accompanied by normal folate levels. If the results of the B12 and folate screening are equivocal, combined use of homocysteine and methylmalonic acid (MMA) levels can differentiate cobalamin from folate deficiency. Most patients with B12 deficiency have elevated levels of homocysteine and MMA, while those with folate deficiency have normal MMA levels [6].
A complete blood count often reveals associated leukopenia and/or thrombocytopenia. Additionally, significant intramedullary hemolysis ("ineffective erythropoiesis"), involving more than 90% of megaloblastic precursors can be detected by a lowered absolute reticulocyte count, increased bilirubin (primarily indirect), decreased haptoglobin, and increased LDH [6]. Catabolism of hemoglobin by the reticuloendothelial cells of the marrow and liver accounts for the increased iron stores and the bilirubinemias [7]. All of these derangements are evident in our patient. Although a bone marrow biopsy is not usually required, blood smear examination is crucial. Neutrophil hypersegmentation is one of the most sensitive and specific signs of megaloblastic anemia. About 98% of patients with megaloblastic anemia have at least one hypersegmented neutrophil per 100 cells examined [1]. Also, a mean corpuscular volume greater than 120 fL is almost always associated with megaloblastic anemia [3]. Megaloblastic anemia is characterized by macro-ovalocytes, in contrast to the round macrocytes usually found in macrocytosis of nonmegaloblastic etiology [1].
A bone marrow biopsy was performed in this patient due to the extreme nature of his pancytopenia. All of the hallmarks of megaloblastic anemia are evident in the specimens. One of the most prominent features is the lack of condensation of nuclear chromatin relative to the degree of hemoglobinization of the cytoplasm. This is most apparent in the more mature erythroid precursors (the last dividing cell class), the early polychromatic megaloblasts [7,8]. In addition, many large metamyelocytes with C-shaped or distorted nuclei are characteristic [8]. As Dr. Antony eloquently describes it, "Most megaloblastic cells are not resting, but vainly engaged in attempting to double their DNA [6]." Due to impaired DNA synthesis, there is frequent arrest in the S phase and lesser degrees of arrest in other phases of the cell cycle. An increased proportion of cells have DNA values between 2N and 4N because of delayed cell division, which is morphologically evident as larger than normal "immature" nuclei with finely particulate chromatin. RNA and protein synthesis is relatively unaffected, so the net result is a megaloblastic cell whose nuclear maturation is arrested while its cytoplasmic maturation proceeds unimpeded. All proliferating cells exhibit similar features, but the changes are most conspicuous in the blood and bone marrow [6]. The large erythroid and myeloid precursors with associated mitotic activity and delayed nuclear maturity can be mistaken for a myelodysplastic syndrome or acute leukemia, if clinical history and laboratory values are unknown [9].
One final point of interest particular to this patient is his history of excessive alcohol intake. Although, in this case, vitamin B12 deficiency is certainly the etiology of the macrocytic anemia, macrocytic red blood cells are a common finding in alcoholism. A diagnosis of alcohol abuse may be considered if macrocytosis is present in the absence of folate deficiency or reticulocytosis. In contrast to our patient, the MCV is usually in the range of 100-110 fL, the macrocytic cells are usually round rather than oval, and there is a concomitant elevation of GGT [6].
Treatment in our patient included a blood transfusion due to the severity of his anemia, then therapy with subcutaneous and oral cyanocobalamin supplements, resulting in stabilization of his laboratory values with some showing rapid normalization. An important aspect of vitamin B12 therapy is that it will need to be continued lifelong [9].
REFERENCES
- Davenport J. Macrocytic anemia. Am Fam Phys. 1996;53:155-162.
- Aster JC. Red blood cell and bleeding disorders. In: Robbins and Cotran Pathologic Basis of Disease. 7th ed. Philadelphia, PA: Elsevier, Inc; 2005:619-659.
- Toh B-H, van Driel IR, Gleeson PA. Mechanisms of disease: pernicious anemia. NEJM. 1997;337:1441-1448.
- Toh B-H, Alderuccio F. Pernicious anemia. Autoimmunity. 2004;37:357-361.
- Schjřnsby H. Vitamin B12 absorption and malabsorption. Gut. 1989;30:1686-1691.
- Antony AC. Megaloblastic anemias. In: Hematology, Basic Principles and Practice. 4th ed. Philadelphia, PA: Elsevier, Inc; 2005:519-556.
- Castle WB. Megaloblastic anemia. Postgrad Med. 1978;64:117-122.
- Wickramasinghe SN. The wide spectrum and unresolved issues of megaloblastic anemia. Sem in Hem. 1999;36:3-18.
- Pruthi RK, Tefferi A. Pernicious anemia revisited. Mayo Clin Proc. 1994;69:144-150.
 Contributed by Siobhan O'Connor, MD and Sandra Kaplan, MD
Contributed by Siobhan O'Connor, MD and Sandra Kaplan, MD



