Final Diagnosis -- Partial Ornithine Transcarbamylase (OTC) Deficiency
FINAL DIAGNOSIS: PARTIAL ORNITHINE TRANSCARBAMYLASE (OTC) DEFICIENCY
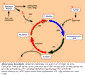 Pathophysiology: OTC deficiency is the most common of the urea cycle disorders (UCD).(1). The urea cycle converts nitrogen from peripheral (muscle) and dietary sources into urea that is water soluble and can be excreted. Figure 2 (2) is a simplified schematic representation of the urea cycle showing how OTC deficiency leads to the impaired condensation of carbamyl phosphate and ornithine to form citrulline. This impairment leads to reduced ammonia incorporation which, in turn, causes symptomatic hyperammonemia and excess of both substrates for the reaction.
Pathophysiology: OTC deficiency is the most common of the urea cycle disorders (UCD).(1). The urea cycle converts nitrogen from peripheral (muscle) and dietary sources into urea that is water soluble and can be excreted. Figure 2 (2) is a simplified schematic representation of the urea cycle showing how OTC deficiency leads to the impaired condensation of carbamyl phosphate and ornithine to form citrulline. This impairment leads to reduced ammonia incorporation which, in turn, causes symptomatic hyperammonemia and excess of both substrates for the reaction.
Under normal circumstances urea cycle occurs in hepatic mitochondria. However when excessive carbamyl phosphate it finds its way into the cytosol where it functions as substrate for the carbamoyl phosphate synthetase (CPS) II reaction. This results in orotic acid, which is a normal intermediate in the very tightly regulated pyrimidine biosynthesis. In OTC deficiency neither conversion of CPS to orotate nor hepatic leakage of ornithine can prevent the rapid development of hyperammonemia (1). The accumulation of ammonia and other metabolites causes. cerebral edema.
OTC is encoded for on the OTC gene located on the X chromosome at band Xp21.1. The nature of mutation in the OTC gene is highly variable with more than 150 mutations, most of which are single-base substitutions, having been reported (3). Most mutations are inherited with a significant rate of spontaneous mutation.
Epidemiology: The estimated incidence of 1:80,000 live births may be an underestimation because late onset cases may go undetected. In older individuals, the initial onset can occur at any age up to 40-50 years and beyond. As an X-linked trait, the mutant OTC gene regularly manifests in hemizygous males (1).
Clinical Presentation: Typically the patient is a term newborn who appears well for the first 24 to 48 hours after birth. It becomes symptomatic after feeding has started because human milk or formula provides a protein load. Initial signs include somnolence and poor feeding, usually followed by vomiting, lethargy, and coma central. Later hyperventilation leading to respiratory alkalosis with abnormal posturing and progressive encephalopathy, hypoventilation and respiratory arrest may follow. Sepsis usually is suspected in a newborn with this presentation. However, the absence of risk factors for sepsis and a non-diagnostic sepsis evaluation prompts consideration of a metabolic disorder (1).
Patients who have partial enzyme deficiencies may have atypical presentations after the newborn period. This delayed presentation is most common with partial OTC deficiency, although it also occurs with partial activity of other urea cycle enzymes. Clinical features in children with atypical presentations include chronic vomiting, developmental delay, seizure disorder, and psychiatric illness. Patients tend to prefer vegetarian diets because dietary protein intake often is associated with migraine like headache. (2)Hyperammonemia may be chronic or recur during metabolic decompensations associated with stress, such as viral illness or childbirth ingestion of drugs that inhibit urea synthesis (eg, valproic acid).
Male hemizygotes usually present in infancy, while female heterozygotes may be totally asymptomatic. Having said that, hemizygous males also may present at any age and well into adulthood without any precedent symptoms or effects, while heterozygous females may be severely affected in childhood. While severity of disease in carrier females can be explained by the nature of the mutation and the random inactivation of the mutant gene, according to the Lyon hypothesis, reasons for late-onset male presentations remain obscure, though some clearly have residual enzyme activity. Reports in the literature show that heterozygous females may also be seriously affected, occasionally suffering mental retardation, and even death from hyperammonemia (4).
Neonatal presentation generally is catastrophic while the late-onset-affected male may suffer a rapid decompensation and demise, similar to the neonatal pattern (1) .
Lab Investigations: In a clinically affected individual, the sine qua non of diagnosis is demonstration of hyperammonemia with associated elevated urinary orotic acid.
The plasma ammonia concentration can be measured in an arterial or venous blood sample. Measurement from a capillary blood sample is not reliable. Blood should be collected in chilled tubes with ammonia-free sodium heparin (green top) or EDTA (purple top), placed on ice, and delivered rapidly to the laboratory. Ammonia levels can be elevated falsely by hemolysis, delayed processing, and exposure to room temperature (2).
If the plasma ammonia concentration is greater than 100 to 150 �mol/L, further testing is performed to establish a diagnosis. Normal values in children older than one month and adults are less than 50 and 30 �mol/L, respectively. Normal values can be higher in newborns than in older children or adults (2).
Initial tests should include arterial pH and carbon dioxide tension, serum lactate, serum glucose, serum electrolytes to calculate the anion gap, plasma amino acids, and urine organic acids and orotic acid. An elevated plasma ammonia concentration combined with normal blood glucose and anion gap strongly suggests a UCD. As other UCDs routinely obtained blood chemistries are not helpful, but a very low BUN may present a diagnostic clue. This should not be interpreted as a substitute for blood ammonia studies because it is not sufficiently reliable. Liver and kidney function typically remain normal unless hypoxia or shock supervenes (1).
Studies that differentiate OTC deficiency from other UCDs include
- Quantitative plasma amino acid analysis showing absent or low citrulline, in association with low arginine and increased glutamine.
- Urine orotic acid is elevated in affected patients and non-symptomatic carriers. If levels are normal in clinically highly suspicious case, allopurinol can be administered to induce orotic acid secretion in the urine of carriers.
- Enzyme analysis of a liver biopsy is usually diagnostic. However OTC activity measured in a liver biopsy may be normal in an affected female, depending upon the pattern of X-inactivation in the liver. It is also important to remember that OTC is a mitochondrial hepatic enzyme and that it is subject to rapid degradation. Therefore any liver biopsy should be done prior to or immediately after death with proper handling of the specimen in order to avoid artefact.
- DNA mutation testing is available and should be performed when this diagnosis is suspected. Detection of a pathogenic mutation in an asymptomatic patient may preclude the need for a liver biopsy to confirm the diagnosis. Molecular methods may also be used to make a prenatal diagnosis. (1)
Differential Diagnosis: The differential diagnosis of neonatal hyperammonemia is limited to UCDs and the unusual transient hyperammonemia of the newborn (THAN). This condition may be distinguished from UCDs by its clinical features including association with lower birth weight and gestational age, earlier presentation of hyperammonemia, and more respiratory distress. Primary genetic causes of hyperammonemia include organic acidemias, fatty acid oxidation defects, and disorders of pyruvate metabolism. These disorders cause hyperammonemia by inhibiting urea cycle enzymes (2).
Causes of hyperammonemia that are not genetic include severe dehydration and liver failure. However, the plasma ammonia level typically is less than 100 to 200 micromol/L in dehydration and returns to normal with volume replacement. Hyperammonemia usually is seen late in the course of severe hepatocellular damage (1).
Treatment: Immediate temporary discontinuation of protein intake in a symptomatic individual is mandatory, with compensatory increases in carbohydrates and lipids in order to offset any catabolic tendency to draw upon muscle amino acids for energy.
In the patient who is comatose with extremely high blood ammonia (in some cases exceeding 2000 mg/dL), rapid reduction can be achieved with hemodialysis.
Intravenously administration of sodium benzoate, arginine, and sodium phenylacetate is important treatment; however, administration of these drugs should be performed in a large medical facility setting with close laboratory monitoring available. IV sodium benzoate and phenylacetate are investigational new drugs. When stabilized the patients are changed to oral citruliline, benzoate and phenylacetate.
As outpatients proper nutrition does not follow the usual nutritional rules and any variations from what is appropriate may result in disaster. This is true throughout life but mostly in the growing infant and adolescent child in whom requirements may fluctuate weekly and must be monitored closely (1).
Patient Counseling: Family pedigree studies in this disease are essential for the following 2 reasons:
- The X-linked nature of the mutation leads to a 1:2 chance of recurrence in any subsequent male conceptus if mother is a carrier. All female siblings of the obligate heterozygous maternal carrier are potential carriers, while male siblings may be at risk for late-onset presentation. Prenatal diagnosis is possible by molecular testsing
- Repeatedly reinforcing the parents in their abilities to perceive early signs of hyperammonemia and to take immediate steps to obtain medical care can be life saving.(5)
Prognosis: Prognosis for older males with initial onset remains unclear because so many remain undiagnosed until very late in the clinical course. However affected male infants with neonatal presentation rarely escape the initial episode with normal mentation. Nonetheless, survival for many years can be achieved with very careful monitoring, use of oral medications and scrupulous dietary attention (5).
REFERENCES:
- Karl S Roth, MD . Ornithine Transcarbamylase Deficiency .emedicine, February 25, 2002
- http://www.uptodateonline.com/application/topic.asp?file=dis_chld/4791&type=P&selectedTitle=3~4
- McCullough BA; Yudkoff M; Batshaw ML; Wilson JM; Raper SE; Tuchman M. Genotype spectrum of ornithine transcarbamylase deficiency: correlation with the clinical and biochemical phenotype. Am J Med Genet 2000 Aug 14;93(4):313-9.
- Arn PH,et al. Hyperammonemia in women with a mutation at the ornithine carbamoyltransferase locus. A cause of postpartum coma. . N Engl J Med 1990 Jun 7;322(23):1652
- Lee, Brendan MD, PhD; Goss, John MD Long-term correction of urea cycle disorders[Proceedings of a Consensus Conference for the Management of Patients With Urea Cycle Disorders] The Journal of Pediatrics. Volume 138(1) Supplement January 2001pp S62-S71
 Contributed by Kudakwashe Chikwava, MD, David N Finegold, MD
Contributed by Kudakwashe Chikwava, MD, David N Finegold, MD




) Pathophysiology: OTC deficiency is the most common of the urea cycle disorders (UCD).(1). The urea cycle converts nitrogen from peripheral (muscle) and dietary sources into urea that is water soluble and can be excreted. Figure 2 (2) is a simplified schematic representation of the urea cycle showing how OTC deficiency leads to the impaired condensation of carbamyl phosphate and ornithine to form citrulline. This impairment leads to reduced ammonia incorporation which, in turn, causes symptomatic hyperammonemia and excess of both substrates for the reaction.
Pathophysiology: OTC deficiency is the most common of the urea cycle disorders (UCD).(1). The urea cycle converts nitrogen from peripheral (muscle) and dietary sources into urea that is water soluble and can be excreted. Figure 2 (2) is a simplified schematic representation of the urea cycle showing how OTC deficiency leads to the impaired condensation of carbamyl phosphate and ornithine to form citrulline. This impairment leads to reduced ammonia incorporation which, in turn, causes symptomatic hyperammonemia and excess of both substrates for the reaction.