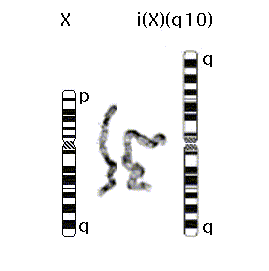
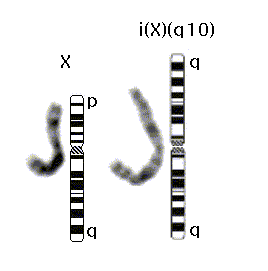
FINAL DIAGNOSIS:
Cytogenetics:
46,X, i(X)(q10)
Abnormal female bone marrow chromosome analysis with one normal X chromosome, and one long-arm isochromosome X derivative. The latter is formed by the centromeric fusion of two copies of the long arm (q) of the X chromosome [ i(X)(q10), also written i(Xq) or iso(Xq) ].
DISCUSSION:
Turner Syndrome was first described by Dr. Henry Turner in 1938 to describe a syndrome of short stature (average adult height 4' 9") and lack of sexual maturation (gonadal agenesis/streak ovaries) in females. Other common physiological findings (>70% of cases) include congenital edema of hands and feet (usually resolved before 2 years of age), broad or "shield-like" chest, unusually shaped or rotated ears, narrow highly arched palate, low posterior hairline, micrognathia, and nail abnormalities. Less common abnormalities (>50% of cases) include "webbed" neck, renal malformations (usually horseshoe kidney), shortened 4th metacarpals or metatarsals, and pigmented nevi. Turner syndrome is not associated with mental retardation (as had been described in older literature), although females with Turner Syndrome may have difficulty in spatial imagery tests and direction-sense skills.
45,X is believed to be the most common embryological chromosomal abnormality , with an estimated incidence of 1-2%. However, the vast majority of these 45,X embryos are aborted spontaneously in the first trimester (estimated 5% prevalence of 45,X in spontaneous abortuses). A small percentage (0.4%) do not abort (for as yet unknown reasons) but develop with relatively minimal physiological sequellae to provide a final incidence of 1/2500 live births. Partial or total deletion of certain non-Lyonized (non-inactivated) genes located on the X (or homologous genes on the Y) chromosome is believed to cause a hypodiploid insufficiency of gene dosage leading to the (partial) expression of Turner phenotype.
There are numerous variant karyotypes seen in Turner Syndrome other than the classic monosomy X ( 45, X ) karyotype. These involve a partial deletion of the second X or Y chromosome such as isochromosome Xq (as seen in this case), or a ring X or Y chromosome. These variant karyotypes are often associated with no features, fewer features, or milder features of Turner Syndrome (as seen in this case). In classic 45,X Turner syndrome, X-linkage studies show that the missing sex chromosome is most frequently of paternal origin (77%) -- likely due to a meiotic nondisjunction event in gametogenesis. However, if the loss of a sex chromosome (or preferential loss of an abnormal sex chromosome derivative such as isochromosome Xq) occurs early in embryogenesis, this may lead to mosaicism (e.g. X/XX, X/iso(Xq), X/XX/XXX, and X/XY mosaicism), which may also be associated with fewer (or no) features of Turner Syndrome.
REFERENCES: