
![]() Contributed by Dimitrios Korentzelos, MD and Steven Dobrowolski, PhD
Contributed by Dimitrios Korentzelos, MD and Steven Dobrowolski, PhD
CLINICAL HISTORY
The patient is a 10-month-old boy born by vaginal delivery to non-consanguineous parents (mother G0 P0, 28 years old) after an uneventful term pregnancy (38 weeks and 5 days). There was no in utero exposure to tobacco, illicit drugs or alcohol, and a standard prenatal care schedule was followed. Although early development after birth was normal, the patient was diagnosed with scoliosis at 3 months of age (~25-30 degrees). At 4 months of age, there were episodes where he appeared to stop breathing. Subsequently, at 6 months of age, he lost the ability to lift his head. A Genetics consult identified failure to thrive, weight loss, and gross motor delay. Initial genetic workup included a single nucleotide polymorphism array, fragile X syndrome assessment, and testing for spinal muscular atrophy, all of which were normal. Autism Spectrum Disorder (ASD) and Intellectual Disability (ID) Comprehensive Panel with automatic reflex to whole exome sequencing was performed but was uninformative, identifying only carrier status for recessive disorders.
Moreover, at 7 months of age, in the course of feeding, gasping was experienced and the patient was taken to a community hospital that identified respiratory acidosis (pH = 6.9, pC02 = 117 mm Hg) with plasma lactate of 5.84 mmol/L, and the child was subsequently intubated. He was started on Keppra (levetiracetam) due to concern for seizures and an EEG showed generalized slowing but no ictal events. Brain magnetic resonance imaging (MRI) was abnormal demonstrating: (a) bilateral, symmetric T2 prolongation within the bilateral basal ganglia, ventromedial thalami, midbrain, tectum and superior cerebellar peduncles concerning for mitochondrial disease, (b) prominence of sulci, extra-axial spaces and ventricular system most consistent with mild diffuse parenchymal volume loss and, (c) limited evaluation of the cervical and thoracic spine due to motion.
As a consequence, metabolic disease work-up was initiated. A set of standard metabolic chemistries, pyruvate, plasma amino acids, and urine organic acids were collected. Chemistries identified elevated lactate (3.8 mmol/L, normal range 0.5-2.2 mmol/L) and pyruvate (0.18 mmol/L, normal range 0.03-0.08 mmol/L). Qualitative urine organic acids showed mild lactic aciduria and significant over-representation of the leucine catabolite 3-hydroxyisovalerate. Glycine conjugates of 3-hydroxyisovalerate were not observed. Based upon results of the urine organic acids, acylcarnitines were assessed leveraging excess plasma and displayed concurrent over-representation of propionylcarnitine (C3) (1.92 μmol/L, normal <1.77 μmol/L) and 3-hydroxyisovalerylcarnitine (C5-OH) (0.16 μmol/L normal 0.0-0.06 μmol/L). Finally, plasma amino acids showed citrulline under-representation (<10 μmol/L, normal 10-70 μmol/L) with normal concentration of alanine (587 μmol/L, normal 148-820 μmol/L). The biochemical results are summarized in Table 1. The sum of biochemical genetic results provided an indication for mitochondrial DNA (mtDNA) sequencing.
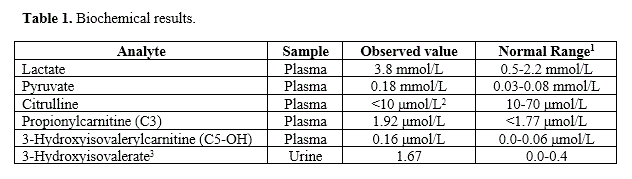
1 Normal range values from the Clinical Biochemical Genetics service of Children's Hospital of Pittsburgh; 2 a modest citrulline (CIT) peak was observed; however, the concentration was below test dynamic range; 3 Urine organic acid testing is qualitative where individual peak areas are measured in respect to the internal standard peak area (which is set to a value of 1.0)
Mitochondrial DNA sequencing identified a homoplastic m.8993T>G (p.L156R) variant which is the most common MT-ATP6 variant and is recognized to be pathological.