FINAL DIAGNOSIS
Leigh syndrome due to a homoplasmic pathogenic variant in the MT-ATP6 gene (m.8993T>G)
DISCUSSION
After integration of the clinical manifestations with biochemical genetic results, the boy was diagnosed with MT-ATP6 deficient Leigh syndrome (infantile necrotizing encephalopathy). This is a rare neurometabolic disorder characterized by progressive central nervous system (brain, spinal cord, and optic nerve) degeneration. Symptoms usually begin between the ages of three months and two years, but some patients might not exhibit signs and symptoms until several years later. Symptoms include loss of previously acquired milestones and motor skills, loss of appetite and failure to thrive, vomiting, irritability and, seizure activity. As the syndrome progresses, generalized weakness, hypotonia, and episodes of lactic acidosis may also appear, thus leading to impairment of respiratory and renal function. Leigh syndrome is a multigenic disorder where deficiencies in >20 different genes are known to be causal. Most genes associated with Leigh syndrome involve mitochondrial energy production, such as deficiency of an enzyme of the mitochondrial respiratory chain complex or the pyruvate dehydrogenase complex. In most cases, Leigh syndrome is inherited as an autosomal recessive trait. However, X-linked recessive and maternal inheritance, due to a mitochondrial DNA mutation, are additional modes of transmission [1]. The latter was true in this case, as the patient featured a homoplasmic pathogenic variant in the MT-ATP6 gene (m.8993T>G). It is presently unknown if the patient's variant is de novo or if inherited from the mother.
This patient is affected with the mitochondrial respiratory chain complex V defect MT-ATP6 deficiency (see Figure 1). Individually, none of the biochemical findings in this patient were overtly severe; however, summation of the biochemical features strongly suggests an MT-ATP6 defect, which mtDNA sequence analysis confirmed. It is possible that defects in MT-ATP6 are under-diagnosed; however, consideration of overall biochemical genetic data may enable more efficient identification of affected patients.
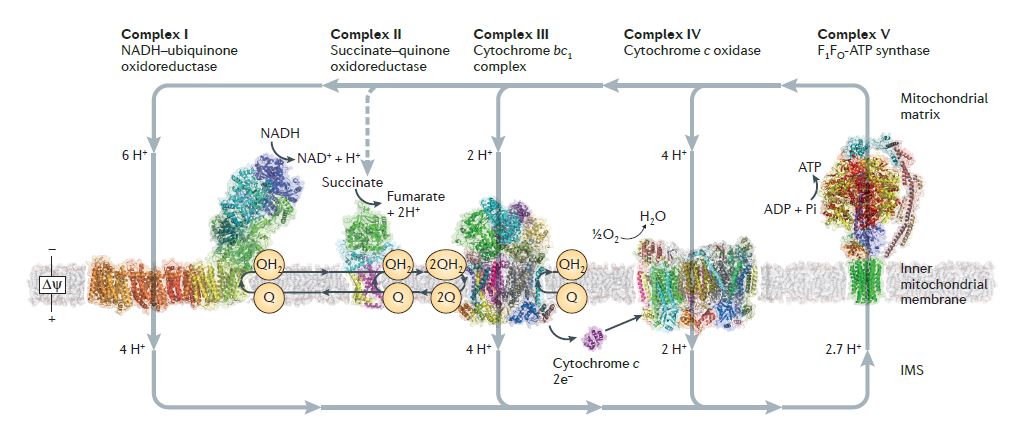
Figure 1. The mitochondrial respiratory chain. (From Sazanov LA. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat Rev Mol Cell Biol. 2015 Jun;16(6):375-88.)
MT-ATP6 gene encodes the mitochondrial membrane ATP synthase (F1F0 ATP synthase, also known as Complex V), which produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the respiratory chain. F-type ATPases consist of two structural domains, F1, containing the extramembranous catalytic core, and F0, containing the membrane proton channel, linked together by a central and a peripheral stalk. During catalysis, ATP synthesis in the catalytic domain of F1 is coupled via a rotary mechanism of the central stalk subunits to proton translocation. This enzyme represents a key component of the proton channel and may also play a direct role in the translocation of protons across the membrane [2].
Disease presentation associated with MT-ATP6 variants includes: Leigh Syndrome, NARP syndrome (neurogenic muscle weakness, ataxia, and retinitis pigmentosa), developmental delay, and other neurologic presentation (dystonia, seizures, etc). The degree of heteroplasmy influences disease presentation [3]. The MT?ATP6 pathogenic variant, m.8993T>G, was characterized three decades ago [4], and has been reported in over 100 patients. A large meta-analysis of 330 patients with Leigh syndrome showed that mutations of the MT-ATP6 gene were the second most frequent (11%) and the m.8993T>G/C MT-ATP6 variant was the most common. Another study demonstrated MT-ATP6 mutations were associated with isolated complex V deficiency [5].
On average, MT?ATP6 variants at position m.8993 appear to result in earlier onset and most severe clinical course, and m.8993T>G causes a more severe disease phenotype compared to m.8993T>C [6]. Biochemical anomalies associated with m.8993T>G include: decreased ATP synthesis (36/38), increased ADP (2/2), normal proton translocation (6/7) & ATP hydrolysis (19/22), decreased holocomplex V assembly (14/18) and, unstable holocomplex V (1/2) [3]. Proposed mechanisms driving these phenomena include anomalous salt bridge formation between subunits a and c causing no rotation of rotor after proton translocation, abnormal holocomplex V production, increased reactive oxygen species generation, decreased ATP synthesis, and abnormal coupling from F0 to F1 [2, 7].
Finally, regarding the phenotype-genotype correlations, a multicenter study by Sofou et al showed that all seven patients with m.8993T>G mutation in their cohort had a severe phenotype with onset by 6 months of age. All patients demonstrated a homoplasmic or >90% heteroplasmic mutation. The aggressive course resulted in 5/7 patients dying by the age of 3 years. The disease onset was characterized by hypotonia (6/7), muscle weakness (4/7) and failure to thrive (4/7). Other clinical manifestations were dyskinesia (5/7), central hypoventilation (4/7), epilepsy (3/7) and nystagmus (3/7). Macular atrophy was seen in one patient. All patients had increased blood and CSF lactate levels. Combined NADH and SDH deficiencies were found in one patient on immunohistochemistry, while another patient had non-specific findings of macrophage infiltration and muscle fiber atrophy. Respiratory chain complex deficiencies were found in four patients; two of them had isolated complex V deficiency and two had generally low complex activities. Brain MRI (6/7) findings were: T2 signal hyperintensity and/or atrophy of the basal ganglia (5/6), midbrain (3/6), cerebellum (3/6), supratentorial white matter (2/6) and cerebral cortex (1/6) [8].
REFERENCES
![]() Contributed by Dimitrios Korentzelos, MD and Steven Dobrowolski, PhD
Contributed by Dimitrios Korentzelos, MD and Steven Dobrowolski, PhD