Brain Pathology Case of the Month - January 2020
 Contributed by Viktoria C. Ruf, MD1, Anne Schöler, PhD2,3, David Capper, MD2,3, Thomas Arzberger, MD1,4, Jochen Herms, MD1, and Ulrich Schüller, MD5, 6, 7
Contributed by Viktoria C. Ruf, MD1, Anne Schöler, PhD2,3, David Capper, MD2,3, Thomas Arzberger, MD1,4, Jochen Herms, MD1, and Ulrich Schüller, MD5, 6, 7
1Center for Neuropathology and Prion Research, Ludwig Maximilians-University Munich, Germany
2 Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin,
and Berlin Institute of Health, Department of Neuropathology, Berlin, Germany
3German Cancer Consortium (DKTK), Partner Site Berlin, German Cancer Research Center (DKFZ), Heidelberg, Germany
4Department of Psychiatry and Psychotherapy, Ludwig-Maximilians-University Munich, Germany
5Department of Pediatric Hematology and Oncology, University Medical Center Hamburg Eppendorf, Germany
6Institute of Neuropathology, University Medical Center Hamburg-Eppendorf, Germany
7Research Institute, Children's Cancer Center Hamburg, Germany
CLINICAL HISTORY
The patient was referred to our hospital for the first time in 2004 as an 8-year-old girl with headaches, nausea, and occasional vomiting. A marginally contrast enhancing mass, completely filling out the fourth ventricle, was detected on brain MRI. Gross total resection was achieved and based on the neuropathological diagnosis the patient underwent radiation and chemotherapy. One year later, progredient contrast enhancement was observed in the right cerebellar hemisphere at the bottom of the fourth ventricle, compatible with progredient residual tumor. These parts were again resected. Further cycles of chemotherapy followed, and the patient recovered well. However, 13 years after the first tumor presentation, the patient started to suffer from headaches and eye movement difficulties. On cranial MRI, a new contrast enhancing mass was found in the posterior fossa. Again, the tumor was resected and referred to histological examination. Shortly thereafter, a myelon-compressing intradural T8-T9 mass was detected and irradiation of the posterior fossa and the thoracic spine was started, when the patient presented with an acute transverse spinal cord syndrome. Resection of this spinal tumor was subsequently performed on an emergency basis. However, based on the rapidly progressive, especially meningeal tumor spread observed on MRI, supportive and palliative care was finally initiated.
MICROSCOPIC PATHOLOGY AND MOLECULAR GENETICS STUDY
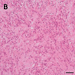
H&E stained sections of the tissue specimen from the first operation showed an exceedingly cellular small blue round cell tumor with densely packed undifferentiated cells and focal rosette-like appearing formations (Fig. 1A). Several mitoses and apoptotic nuclei were observed, but necrosis or vascular proliferation were not detected. GFAP was only focally expressed, whereas weak to moderate synaptophysin staining was present throughout the tumor. The Ki-67 proliferating index was close to 90 % (data not shown).



The second tumor specimen demonstrated a glial tumor of low to moderate cell density, where the tumor cells were grouped pseudo-rosette-like around hyalinized blood vessels (Fig. 1B). Mitoses and necrosis were not observed. The tumor cells stained positive for GFAP and S-100, whereas no positivity for synaptophysin was found. Only about 3 % of the tumor cell nuclei expressed Ki-67 (data not shown).
H&E sections of the third resection showed a pleomorphic, moderately cellular glial tumor with geographic necrosis and prominent hyalinized microvascular proliferation (Fig. 1C). Rosette-like formations were not observed. Reticulin fibers were limited to vascular structures. Olig2 and MAP2 were strongly expressed throughout the tumor tissue, while GFAP showed in some areas only weak to moderate staining. Pronounced nuclear accumulation of p53 was found in a number of tumor cells. Mutated H3 (K27M) was not detected, but trimethylation of Histone H3 at position K27 was lost. Up to 60 % of the nuclei were Ki67-positive. Sequencing of IDH1/2, TERT promoter regions, and the H3-genes revealed no abnormalities.
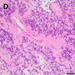
Sections of the fourth, spinal resection revealed a moderately pleomorphic, remarkably apoptotic and necrotic glial tumor, in which the tumor cells were arranged in a pseudo-rosette-like manner around blood vessels (Fig. 1D). A fraction of the tumor cells strongly expressed GFAP and MAP2, but were negative for Olig2. Trimethylation of Histone H3 at position K27 was lost. The Ki67 proliferating index was focally up to 20 %.
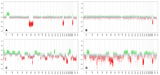
Global Methylation analyses were finally obtained from all four tumor specimens using 450K or 850K bead chip arrays and results were analyzed using a brain tumor classifier that included several thousands of reference brain tumor data (Capper et al., in press). As shown in Figure 1E, samples from the first (1), the second (2), and the fourth (4) operation demonstrated high similarity to posterior fossa ependymomas, group A. In contrast, tissue from the third (3) operation fell very close to diffuse midline glioma using t-distributed stochastic neighbor embedding (t-SNE). While the copy number variation profiles (CNV) of the first tumor sample showed only few chromosomal aberrations and the chromosomes from the second resection seemed to be nearly balanced (Fig. 1F-a, b), the third and the fourth tumor samples exhibited a quite complex pattern of chromosomal aberrations (Fig. 1F-c, d).
Finally, neither of the characteristically glioma-associated mutations of IDH1/2, BRAF, H3 or the TERT promoter sequence could be detected at least in the two latest received specimens. What are your diagnoses?
FINAL DIAGNOSIS
)
)
)
)
)
)