Brain Pathology Case of the Month - August 2017
 Contributed by Katherine E. Schwetye, MD, PhD1, Karen Gauvain, MD2, David Rodriguez, MD3, Catherine Cottrell, PhD1,
Contributed by Katherine E. Schwetye, MD, PhD1, Karen Gauvain, MD2, David Rodriguez, MD3, Catherine Cottrell, PhD1,
David D. Limbrick, Jr., MD, PhD,4 Robert E. Schmidt, MD, PhD1, Sonika Dahiya, MD1
1Department of Pathology and Immunology
2Department of Pediatrics, Division of Hematology-Oncology
3Mallinckrodt Institute of Radiology, Department of Neuroradiology
4Department of Neurological Surgery, Washington University School of Medicine, St. Louis, MO, USA
CLINICAL HISTORY
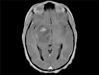
An 8-year-old, previously healthy girl presented with a 3-month history of intermittent, progressive night-time and early morning headaches and vomiting, and a three week history of left-sided facial weakness and mild left hemiparesis. Magnetic resonance imaging showed a 5.6 x 4.3 x 4.5 cm T1 hypointense, T2/FLAIR hyperintense mass containing several scattered foci of contrast enhancement centered within the right basal ganglia and medial temporal lobe (Fig. 1A). The mass demonstrated restricted diffusion, and contained no hemorrhage, necrosis, calcification, or surrounding vasogenic edema. The presence of restricted diffusion and heterogenous enhancement on imaging suggested a highly cellular tumor. Given the young age of this patient, the radiological differential considerations included primitive neuroectodermal tumor (PNET) and anaplastic ependymoma especially because of the lack of surrounding vasogenic edema. A stereotactic needle biopsy was performed. The patient was treated with conformal radiation therapy and concurrent chemotherapy with temozolomide, followed by temozolomide and lomustine according to the Children's Oncology Group ACNS0423 protocol. She had clinical and radiographic response to treatment until 7 months following diagnosis, when her hemiparesis returned and she developed vision loss. MRI revealed marked interval progression of her disease with extension into her right optic tract, optic chiasm, and brainstem. She was then treated with bevacizumab and irinotecan. Two months later, she developed further progression with new subependymal spread and extension into the left cerebrum and brainstem. Her therapy was discontinued and she expired 12 months from the date of her initial diagnosis.
MICROSCOPIC AND MOLECULAR PATHOLOGY






Histological examination of the biopsy material showed a high-grade diffuse glioma. Through-out, the tumor cells showed a rather uniform appearance (Fig. 1B). Other notable features included the focal presence of myxoid/mucinous background, delicate thin-walled capillaries, and pericellular clearing with absence of definite microvascular proliferation and/or necrosis. While the tumor cells displayed deceptively monomorphic appearance on low-power, there was significant pleomorphism and brisk mitotic activity on high-power examination (Fig. 1C) which was reflected by its overall high Ki-67 proliferation as well. The hyperchromatic tumor nuclei had irregular nuclear membranes. Immunohistochemistry demonstrated a majority of tumor cells to be positive for glial fibrillary acidic protein (GFAP) (Fig. 1D), with retained ATRX expression (Fig. 1E), and non-reactivity for p53, mutant IDH1 (R132H), and synaptophysin. Fluorescence in situ hybridization (FISH) studies showed normal dosages of chromosomes 1 and 19 and no evidence of EGFR amplification although there was polysomy of chromosome 7. Loss of PTEN by monosomy of chromosome10/10q was also present (Fig. 1F). Additionally, molecular analysis was performed using a next-generation sequencing platform to analyze a select panel of genes, including IDH1, IDH2, TP53, BRAF, EGFR, PTEN, ATRX, PDGFRA, and H3F3. This work-up revealed mutation in the H3F3 gene due to an amino acid change (K27M) which has frequently been described in pediatric high-grade gliomas. An immunohistochemical stain for this mutant protein (H3K27M) was also positive in tumor cells in concordance with the sequencing results (Fig. 1G). What is your diagnosis?
FINAL DIAGNOSIS
)
)
)
)
)
)
)