![]() Contributed by Robert M. Sargis, MD, PhD1; Robert L. Wollmann, MD, PhD2; and Peter Pytel, MD2
Contributed by Robert M. Sargis, MD, PhD1; Robert L. Wollmann, MD, PhD2; and Peter Pytel, MD2
![]() 1University of Chicago, Department of Medicine, Section of Endocrinology; 2University of Chicago, Department of Pathology
1University of Chicago, Department of Medicine, Section of Endocrinology; 2University of Chicago, Department of Pathology
CLINICAL HISTORY AND IMAGING STUDIES
A 59 year-old man with no significant past medical history presented to our neurosurgery clinic with progressive vision loss. He initially noted partial loss of vision in his right eye four years prior to being seen at our institution; this subsequently progressed to complete unilateral blindness over the following year and a half. During this time he had seen a physician at an outside hospital with an unclear work-up and no treatment. His visual loss was then stable for two and a half years until he noticed transient loss of vision in his left eye at which point he was referred to our institution. Magnetic resonance imaging of his brain revealed a 3.3 cm x 3.3 cm x 3.4 cm suprasellar mass (Figure A). Transcranial resection was recommended; however, the patient initially deferred treatment. Two months later he presented to our emergency department with further visual disturbances in his left eye including longer periods of blindness and episodes of color distortion. He was admitted to our neurosurgical service for surgical resection of the mass.
)
On physical exam the patient's blood pressure was 154/64 and his heart rate was in the high 40s, with an EKG showing sinus bradycardia. The patient denied symptoms of thyroid dysfunction, changes in his physical appearance, nipple discharge, and sexual dysfunction. He had no vision in his right eye and temporal hemianopsia on the left; his gaze was disconjugate with lateral deviation of his right eye. The rest of his neurological examination was within normal limits. Pertinent negatives included the absence of gynecomastia, galactorrhea, and Cushingoid or acromegalic features. His testes were of normal size and consistency, and he had a normal body hair distribution.
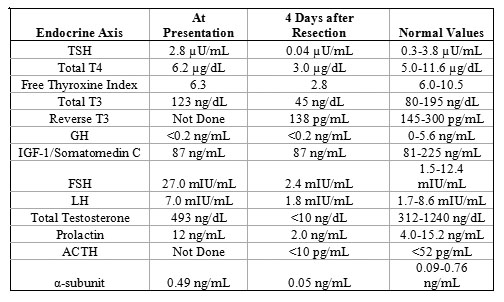
Endocrine evaluation revealed the results presented in Table 1. With the exception of the elevated follicle stimulating hormone (FSH), all of the patient's laboratory results were within normal limits. Dilution of the patient's prolactin sample was done to ensure that there was not false normalization with our sandwich enzyme-linked immunosorbent assay; the diluted samples were all within normal limits (results not shown).
The patient was treated initially with high-dose methylprednisolone with subsequent resolution of his left-sided visual disturbances. On admission day four, he underwent subtotal transcranial resection of the suprasellar mass. Post-operatively the patient's course was complicated by transient diabetes insipidus treated with several doses of desmopressin. Results of the endocrine evaluation four days after surgery are shown in Table 1. Therapy was initiated with levothyroxine, hydrocortisone, and testosterone; he was discharged 17 days after his resection.
NEUROPATHOLOGICAL FINDINGS
The majority of the tumor showed typical pituitary adenoma morphology with vaguely nested sheets of monomorphic cells. These were found to exhibit immunoreactivity for synaptophysin, FSH, and luteinizing hormone (LH) as well as focal reactivity for thyroid stimulating hormone (TSH). The tumor cells were negative for adrenocorticotropic hormone (ACTH), growth hormone (GH), and prolactin. MIB-1 staining for the proliferative marker Ki-67 labeled less than 2% of cells.
)
)
)
)
)
Scattered throughout the adenoma were small nests of epithelial cells with squamoid differentiation that showed peripheral palisading of the nuclei and focal stellate reticular appearance (Figure B and C). These nests were immunoreactive for high molecular weight cytokeratin (HMWK, Figure D) and p63 (Figure E). Some of the basal cells within these nests showed proliferative activity as assessed by labeling with the MIB-1 antibody (Figure F).