Brain Pathology Case of the Month - February 2007
 Contributed by Werner Stenzel1, Marek Franitza1, Albert J. Becker2, Manuel Montesinos-Rongen1
Contributed by Werner Stenzel1, Marek Franitza1, Albert J. Becker2, Manuel Montesinos-Rongen1
Mario Löhr3, Jin-Yul Lee3, Gabriele Röhn3, Hrvoje Miletic1, and Martina Deckert1
1 Department of Neuropathology, University of Cologne, Cologne, Germany
2 Department of Neuropathology, University of Bonn, Bonn, Germany
3 Department of Neurosurgery, University of Cologne, Cologne, Germany
CLINICA: HISTORY:
A 30-years old male patient had suffered from childhood epilepsy at the age of three years. He presented with multiple cutaneous angiofibromas, bilateral renal angiomyolipomas and a tumor of the liver that had not been biopsied. His family history was unremarkable; in particular, TSC history was negative.
Having complained of increasingly severe headache over six months, while neurological examination, laboratory studies and EEG were normal, he was operated for a tumor mass in the right lateral ventricle. Macroscopically, the tumor was entirely removed. Physical examination excluded any postoperative neurological deficit. Although follow-up cranial MRI four months later did not show any evidence for residual tumor, the patient developed an early relapse five months after surgery. He was treated postoperatively by external irradiation with 50.4 Gy. Six months thereafter he complained of imbalance and gait difficulties, and MRI of the spinal axis showed a tumor of the cervical spinal cord at C6/C7, which was surgically removed. The patient died seven months later. Autopsy and genetic testing of his family was refused.
RADIOLOGY:
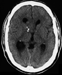
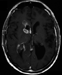
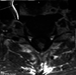
Preoperative CCT of the primary tumor showed an isodense tumor with small calcifications in the right anterior horn of lateral ventricle obstructing the foramina of Monro (Fig. 1a). The T/1 Gd enhanced MRI, showed a local relapse with additional subependymal growth pattern and a second lesion obstructing the trigone of the right lateral ventricle (Fig. 1b). Transversal Gd-enhanced T1/MR imaging demonstrated an enhancing cervical spinal lesion (Fig. 1c).
MICROSCOPIC FINDINGS:

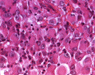
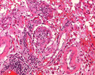
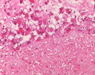
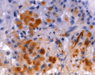



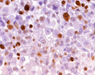

Neuropathological examination of all three specimens on paraffin embedded and cryostat sections disclosed a tumor composed of polymorphous, large, stunted cells (Fig. 2.a), sometimes clustering around blood vessels with the formation of perivascular pseudorosettes. In addition, huge ganglioid cells and elongated fibrillary cells were obvious (Fig. 2.e). Furthermore elevated mitotic activity (12/10 high power fields; HPF = 0.16mm2) and necrosis were seen. The intracerebral recurrence (Fig. 2.b) displayed markedly more mitoses (21/10 HPF) than the primary tumor, vessel proliferation (Fig. 2.c) and necrosis (Fig. 2.d). Immunohistochemistry exhibited a mixed glio-neuronal phenotype of the primary tumor with positive staining of the tumor cells for GFAP (Fig. 2.e), vimentin, S100 protein, synaptophysin, neurofilament, and NSE, while the p53 protein was not expressed (data not shown). The spinal metastasis (Fig. 2.f) had lost immunoreactivity for glial (GFAP) (Fig. 2.g) and neuronal markers (NFP, synaptophysin, NSE). The fraction of MIB-1 positive tumor cells paralleled the mitotic activity of the tumors (primary tumor: 20 % MIB-1 positive & 12 mitoses/10 HPF; recurrence: 40 % MIB-1 positive & 21 mitoses /10 HPF; spinal metastasis: 5 % MIB-1 positive & 6 mitoses/10 HPF; Fig. 2.h, 2.j).
FINAL DIAGNOSIS
)
)
)
)
)
)
)
)
)
)
)
)
)