FINAL DIAGNOSIS
Introduction:
Hereditary elliptocytosis (HE) and its variants are congenital hemolytic disorders in which erythrocytes are either elongated into a cigar or oval shape or are poikilocytic and bizarrely shaped (1). The presence of at least 25% of cells which have an elliptocyte morphology has been suggested as a criterion to diagnose hereditary elliptocytosis. However, the prevalence of elliptocyte cells in the peripheral blood smear can vary from 0-100%. Inheritance of hereditary elliptocytosis is autosomal dominant. However, de novo mutations have been reported in rare cases (1).
The frequency of this disorder ranges from 1/5000 to 1/10,000 population among Caucasians. Worldwide, the incidence is estimated to be 1/2000-4000 individuals (3). It is more common in regions where malaria is endemic; the prevalence in West Africa approaches 6/1000 (3). The true incidence is unknown because the clinical severity of hereditary elliptocytosis is heterogeneous, and many patients are asymptomatic. The incidence of an elliptocytosis variant known as Southeast Asian ovalocytosis ranges from 5-25% in Melanesia, Philippines, Indonesia, and southern Thailand (4). The majority of patients with hereditary elliptocytosis are asymptomatic, and the diagnosis is made incidentally.
Pathophysiology:
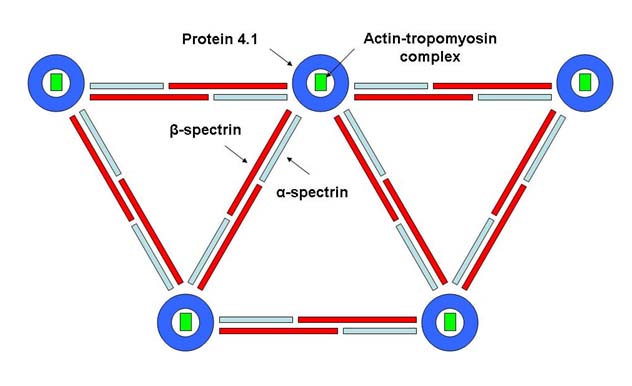
Hereditary elliptocytosis and its related variants are caused by mutations that disrupt the RBC cytoskeleton, a multiprotein complex responsible for the elasticity and durability of the circulating erythrocytes. Figure 4 indicates with a schematic the organization of the sub-membrane protein structure network in RBCs. Spectrin tetramers form a large part of the cytoskeletal framework and are composed of heterodimers of alpha and beta subunits. These are tethered to the plasma membrane proteins AE1 (band 3) and glycophorin C through the ankyrin/protein 4.2 complex and through protein 4.1R and its associated actin filaments.
Mutations that disrupt the formation of spectrin tetramers result in hereditary elliptocytosis. Most of the elliptogenic mutations affect the alpha and beta structures of spectrin molecules which comprise a major part of the sum-membrane meshwork. Approximately 65% of cases of hereditary elliptocytosis are the result of mutations of alpha spectrin, 30% are the result of mutations of beta spectrin, and 5% are the result of mutations of protein 4.1 (2). RBC precursors in common hereditary elliptocytosis are normally round but become more elliptical as they age. The mutation of a gene on the X chromosome can give rise to elliptocytosis associated with Alport's syndrome. Elliptocytes and poikilocytes are postulated to be permanently stabilized in their abnormal shape because the weakened skeletal interactions facilitate skeletal reorganization after prolonged or repetitive cellular deformation. This may result in hemolytic anemia with RBC fragmentation. Splenic sequestration is the dominant cause of the decreased survival of these abnormal red cells.
Clinical presentation
Most patients are clinically asymptomatic, and the diagnosis is usually made incidentally when a blood smear is examined. Asymptomatic patients are heterozygous for the disease and are classified as having mild or common hereditary elliptocytosis. Approximately 10% of patients have moderate to severe anemia, with intermittent episodes of acute hemolysis with jaundice and splenomegaly. There are three main forms of hereditary elliptocytosis (1):
Severe hereditary elliptocytosis and hereditary pyropoikilocytosis (HPP) - In general, patients with homozygous hereditary elliptocytosis (HPP) have symptomatic hemolytic anemia that requires transfusion support and eventual splenectomy. This is of special concern in pregnant women who carry the gene and may transfer it to their offspring. Newborns thus should be monitored for development of symptoms of HPP in the neonatal period. Patients with HPP typically present in the early newborn period with severe hemolytic anemia, RBC fragmentation, poikilocytosis, elliptocytosis, and microspherocytosis, as evident on peripheral blood smears (2). Neonatal hyperbilirubinemia and severe anemia in the first few months of life are the typical presenting signs. Complications of severe anemia, including splenomegaly, growth retardation, frontal bossing, and early gallbladder disease, are common. More severe cases may result from coinheritance of a typical hereditary elliptocytosis mutation and a relatively common but weak alpha gene allele that results in clinically apparent hemolytic anemia. There may be homozygosity or compound heterozygosity for a mutant spectrin affecting the dimmer self-assembly in the cells (2). HPP in infants can evolve into more typical mild hereditary elliptocytosis with concomitant improvement of symptoms and anemia.
The mortality and morbidity in these disorders depends on the frequency and degree of hemolytic anemia. The clinical phenotype ranges from asymptomatic carrier status to severe transfusion-dependent, and even fatal, hemolytic anemia. Individuals with chronic hemolysis may have complications such as jaundice, splenomegaly, and early gallbladder disease. Mortality is rare. Most patients have normal physical examination findings, but should be evaluated for signs of cardiovascular compromise. Patients undergoing hemolysis may have pallor, jaundice, or splenomegaly.
Laboratory investigations and diagnosis
The primary approach to diagnosis is based on clinical suspicion. As mentioned before, cases are usually discovered during a routine peripheral blood examination for other causes. These disorders are suspected in patients with unexplained hemolysis, particularly if splenomegaly or a family history are present, or abnormal RBC indices are identified. Because RBCs are spheroidal and the MCV is normal, the mean corpuscular diameter is below normal, and RBCs resemble microspherocytes. MCHC is increased. Reticulocytosis of 15 to 30% and leukocytosis are common.
If these disorders are suspected, the RBC osmotic fragility test (which mixes RBCs with varying concentrations of saline) and the RBC autohemolysis test (measuring the amount of spontaneous hemolysis after 48 h of sterile incubation) can be performed. In addition, direct antiglobulin (Coombs') test may be done to rule out spherocytosis due to autoimmune hemolytic anemia. RBC fragility is characteristically increased, but in mild cases, it may be normal. RBC autohemolysis is increased and can be corrected by the addition of glucose.
The confirmatory testing for the presence of hereditary elliptocytosis is based on molecular genetic testing for the presence of mutations in the specific protein molecules of the RBCs. Genetic counseling should be offered to parents with this condition to explain the risks regarding the transmission of the condition and especially regarding the potential dangers of HPP in the newborn.
Differential Diagnosis:
In addition to the hereditary causes of elliptocytosis, it is important to consider the non-hereditary or acquired causes of elliptocytosis.2 There are a multitude of acquired hematological disorders which can lead to elliptocyte RBC forms in the peripheral blood smear. These include iron deficiency anemia, thalassemia, megaloblastic anemia, myelofibrosis, myelophthisic anemia, myelodysplastic syndrome and pyruvate kinase deficiency. These causes must be ruled out before investigating hereditary causes of elliptocytosis.
Patient follow-up:
The peripheral blood morphologic findings are compatible with a diagnosis of hereditary elliptocytosis. The patient refused any form of further follow-up testing to confirm the diagnosis and to establish the exact nature of the mutations involved in the genesis of the abnormal RBC phenotype. She was provided however with appropriate genetic counseling.
REFERENCES
![]() Contributed by Ramachandra Gullapalli, MD, PhD, Anne Shaheen MD and Lydia Contis, MD
Contributed by Ramachandra Gullapalli, MD, PhD, Anne Shaheen MD and Lydia Contis, MD