FINAL DIAGNOSIS
Histologic transformation to small cell lung carcinoma from an EGFR-mutated lung adenocarcinoma, a mechanism of resistance to EGFR tyrosine kinase inhibitor treatment.
DISCUSSION
Lung cancer is one of the deadliest forms of cancer, accounting for 21.7% of all cancer deaths while making up only 12.4% of new cancer cases in 2021.[1] Non-small cell carcinoma (adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma) and small cell carcinoma account for the majority of types of lung cancer. Small cell lung carcinoma (SCLC) is often treated initially with systemic chemotherapy due to its aggressiveness and advanced stage at detection. It is strongly associated with smoking history with less than 10% 5-year survival rate, largely due to advanced stage at presentation and lack of significant advances in therapeutics; although this may be changing with the emergence of immunotherapy.[2] In contrast, because there are multiple FDA approved agents available for patients with advanced stage adenocarcinoma based on the oncogenic profile of a lung adenocarcinoma, molecular profiling of lung adenocarcinoma, especially the more advanced staged adenocarcinomas, are standard of care. Guidelines now suggest that extensive immunohistochemical workup should be avoided in small biopsies to save material for molecular studies.[3] This case demonstrates the interesting relationship between histological findings, molecular data, and therapy regimens.
Lung adenocarcinomas (LUAD) in cytologic preparations have an appearance similar to many other types of adenocarcinomas. The cells form three-dimensional clusters with eccentrically placed, irregular nuclei, prominent nucleoli, and mucin vacuoles.[2] On biopsy specimens, the cells of non-mucinous adenocarcinoma can display lepidic, acinar, papillary, micropapillary, cribriform, and solid architectural patterns, often with a combination of patterns present. Evidence of invasion includes non-lepidic patterns, invasive tumor cells within stroma, vascular invasion, pleural invasion, or spread through airspace. Tumors measuring greater than 2.5cm, containing solid architectural pattern, or containing micropapillary architectural pattern generally have a worse prognosis.[4] Evaluation using a minimal number of immunohistochemical stains, such as only p40 and TTF-1, is suggested to demonstrate that the tumor cells are of lung origin/adenocarcinoma (e.g. TTF-1 positive) and are non-squamous (e.g. p40 negative), while preserving material for molecular studies.[3]
Molecular profiling is essential for advanced stage LUAD treatment decision planning since the choice of chemotherapy, immunotherapy, or targeted therapy as a first line systemic treatment can be based on the presence of driver oncogene molecular alterations that have FDA approved inhibitors; such oncogenes in LUAD include EGFR, ALK, ROS1, BRAF, KRAS, MET, and NTRK1/2/3. Activating mutations in EGFR are most commonly p.L858R or exon 19 deletions, and they are seen in up to 15% of LUAD (more frequent in never-smokers).[5] Gene rearrangements of ALK, ROS1, or RET, which can all be detected by next generation sequencing or FISH studies, are also responsive to select tyrosine kinase inhibitors (TKIs), based on the affected gene. A specific mutation in BRAF, p.V600E, is responsive to combinations of BRAF and MEK inhibitors (such as dabrafenib and trametinib).[6] Mutations in KRAS generally suggest a poor prognosis and less responsiveness to TKI therapy, although a specific p.G12C mutation may be treated with sotorasib.[7] Mutations that cause exon 14 skipping in the MET gene are responsive to a MET TKI, like capmatinib.[8] Finally, gene fusions containing NTRK1/2/3 are rare in LUAD, but are associated with responsiveness to TRK inhibitors (larotrectinib, entrectinib).[9]
The patient in this case had a LUAD with a common type of EGFR mutation, an exon 19 deletion, which was treated with osimertinib, a third generation EGFR TKI. Osimertinib treatment is associated with improved survival benefits in the setting of EGFR-mutated LUAD in both EGFR treatment na�ve patients and patients previously treated with EGFR TKI who have developed p.T790M resistance mutations.[10, 11] Treatment resistance to first generation EGFR TKIs are most commonly associated with additional mutations in EGFR (e.g. p.T790M for first generation TKI, p.C797S for osimertinib), bypass signal activation such as gene copy amplification of the MET gene, and histologic transformation to small cell carcinoma.[12] For osimertinib, histologic transformation to squamous cell carcinoma occurs as well.[13,14]
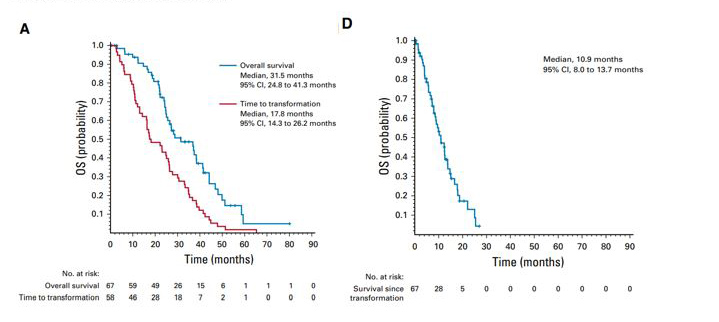
Up to 10% of EGFR-mutant LUAD undergo transformation to SCLC for first generation EGFR TKIs and is also an important mechanism of resistance to osimertinib. In patients who had histologic transformation to SCLC in the setting of first generative EGFR TKI therapy, the median time to transformation in one study was 17.8 months.[15] That study showed that transformed SCLC has behavior clinically similar to those de novo SCLC (non-EGFR mutated), with transient therapy response, frequent CNS metastasis, and short overall survival time (Figure 3). [15]
SCLC often has TP53 and RB1 genomic alterations, among many other types of driver mutations. While this case demonstrated a tumor that acquired those mutations at the same time as the histologic transformation, only approximately 25% of LUAD that have EGFR, RB1, and TP53 mutations underwent transformation in one retrospective study (Figure 4).[16]
Figure 3: Survival curves of time to transformation and overall survival in SCLC transformed from EGFR-mutated LUAD.
From: Marcoux N, et al. J Clin Oncol. 2019.[15] Survival curves demonstrating the time to transformation (A) and overall survival post-transformation (D) in a retrospective study of 67 cases of EGFR-mutated LUAD that underwent transformation into SCLC.
Small cell carcinoma has a unique cytologic presentation in which cells are small with minimal cytoplasm, molding and nuclei with "powdery" chromatin without distinct nucleoli. Mitoses should be readily apparent. There is often cellular debris, crush artifact, and some paranuclear blue bodies. The cytologic preparation in Figure 2 of this case demonstrates powdery chromatin, some nuclear molding, and crush artifact. SCLC must be differentiated from carcinoid tumor, atypical carcinoid tumor, lymphoid aggregations, reserve cell hyperplasia, and small round blue cell tumors.[2] Immunohistochemical stains on SCLC show a rim-and-dot-type pattern for AE1/AE3 and CAM5.2, positivity for neuroendocrine markers (synaptophysin, chromogranin, and CD56), positivity for TTF-1, negativity for Napsin A, and negativity for p63 or p40. Ki-67 proliferative index is usually very high (65-100%). Consistent with the molecular findings in SCLC, IHC usually shows loss of RB1 and aberrant p53 expression.[4]
Figure 4: Histologic patterns and additional mutations in lung cancers containing EGFR, RB1, and TP53 mutations.
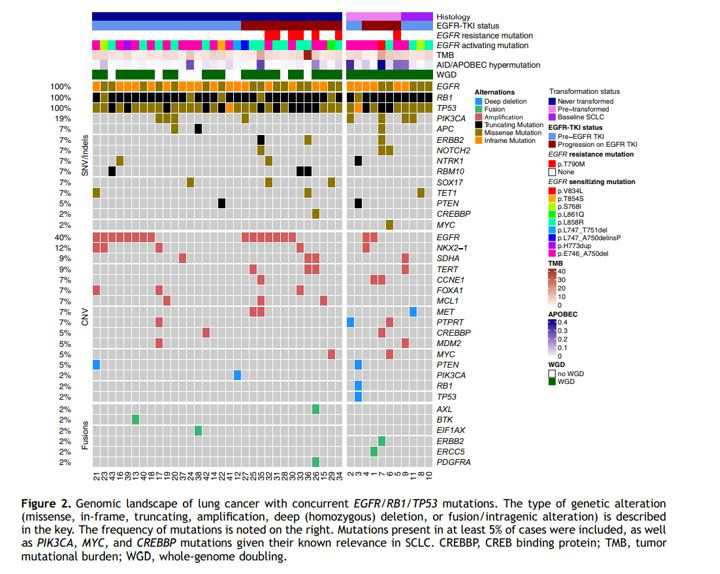
From: Offin M, et al. J Thorac Oncol. 2019. [16]
This case demonstrates an EGFR-mutated LUAD transformation to SCLC, likely in response to TKI therapy. It highlights the importance of correlations between histologic findings and prognosis, molecular findings and treatment, and treatment-related resistance mechanisms both histologically and through molecular mechanisms.
REFERENCES
![]() Contributed by Jeff Kleinberger, MD, PhD and Gabriel Sica, MD, PhD
Contributed by Jeff Kleinberger, MD, PhD and Gabriel Sica, MD, PhD