
![]() Contributed by Daniel Marker, MD, PhD
Contributed by Daniel Marker, MD, PhD
CLINICAL HISTORY
A young previously healthy child with no significant medical history presented to their primary care physician with a history of intermittent headaches and fevers for the past few months. The headaches had no predictable pattern and no positional triggers or intensity change. The patient was initially treated with oral antibiotics, which failed to improve their symptoms. They then developed gait disturbances and visual changes, prompting imaging studies (Figures 1 and 2).
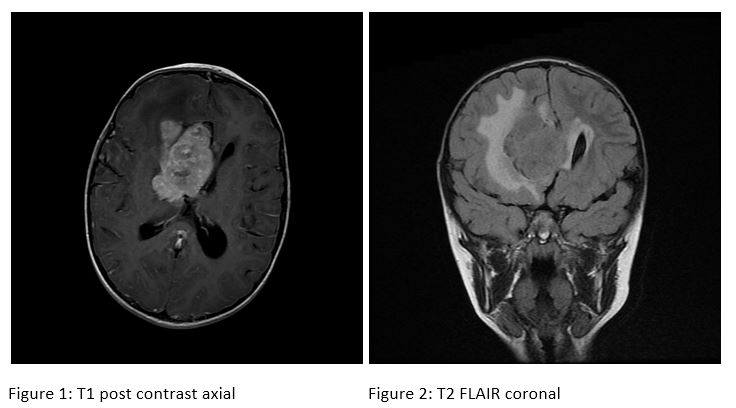
MRI of the head revealed a 5.8 x 4.2 x 4.1 cm heterogenously enhancing lobulated mass in the right lateral ventricle that involved the right frontal lobe periventricular white matter and abutted the right thalamus. There was extensive surrounding vasogenic edema and partial obstruction of the foramen of Monroe resulting in mild lateral ventriculomegaly. The patient was referred to Children's Hospital of Pittsburgh and taken to the OR for tumor resection.
MICROSCOPIC FINDINGS
Intraoperative smear:
http://image.upmc.edu:8080/NeuroPathology/PediatricTumors/PediatricTumor5/PT.67a.svs/view.apml?
The intraoperative smear showed huge bizarre multinucleated cells along with a smaller tumor cell population. Mitotic figures were present and the dirty background was concerning for necrosis. The differential diagnosis included high-grade primary brain tumor versus metastatic poorly differentiated tumor from unknown primary. The intraoperative diagnosis was, "morphologically bizarre tumor, defer to permanents."
Permanent H&E:
http://image.upmc.edu:8080/NeuroPathology/PediatricTumors/PediatricTumor5/PT.67b.svs/view.apml?
Permanent sections showed the same huge bizarre tumor cell population with small cell component. Extensive necrosis was present. No definitive parenchymal infiltration was identified. The tumor cells showed patchy positivity for GFAP and were negative for Olig2, TTF1, synaptophysin, Cam5.2, EMA, Sox-10, and Gata3, although the interpretation of the immunostains was limited by extensive cautery artifact. TP53 was suspicious for clonal loss. The Ki67 proliferation index was approximately 30%. Molecular analysis of the tumor was performed and identified stop-gain mutations in TP53, RB1, and TSC2.