
![]() Gaurav Kattel, MBBS, MD and Steven F. Dobrowolski, PhD
Gaurav Kattel, MBBS, MD and Steven F. Dobrowolski, PhD
CLINICAL HISTORY
A 40-year-old male presented with progressive blurry vision and a restricted peripheral vision. He also described seeing "tiny raindrops" in the frontal vision field.
His vision problems began in childhood when, despite glasses, his vision was not comparable to peers. Night blindness was a life-long persistent element of his presentation. He had a recent history of cataract surgery followed by a YAG capsulotomy that did not help with his vision issues.
As a member of the Amish community, there was a history of consanguinity. A female sibling presented with similar, albeit milder, visual impairment. A life-long learning disability and nystagmus were reported. Alcohol use was denied.
His visual acuity was 20/400 OD (Oculus Dexter) and 20/300 OS (Oculus Sinister). Dilated fundus exam showed waxy pallor of the discs, macular atrophy, attenuated vessels, and peripheral bone spicule pigmentation. Ocular coherence tomography (OCT) demonstrated retinal thinning, central atrophy, and choroidal thinning. Ocular field testing showed tubular vision with severe constriction.
Based on family history, clinical phenotypes, and imaging, he was referred for a genetics consult.
Genetic testing results:
Primary variant:
A novel homozygous SRD5A3 c.176dup, p.(Pro60Alafs*110), variant was identified and owing to frameshift was categorized as likely pathogenic.
Carbohydrate deficient transferrin (CDT) results:
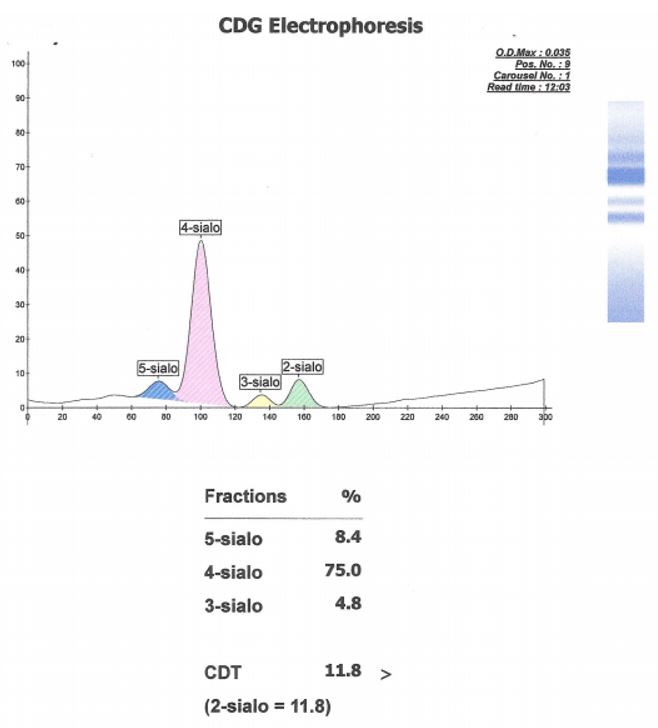
Capillary electrophoresis of serum sialotransferrins revealed an abnormal pattern. While high-order silated species (5-sialo, 4-sialo) retained normal representation, disialo species were in excess of trisialo species. This pattern suggested a glycosylation deficiency consistent with the homozygous SRD5A3 mutation. These results indicated SRD5A3-CDG, also known as CDG-1q.