FINAL DIAGNOSIS:
KIDNEY, LEFF, PERCUTANEOUS NEEDLE BIOPSY:
DISCUSSION
Dense deposit disease (DDD), also known as membranoproliferative glomerulonephritis type II (MPGN-II), is a clinicopathologic entity associated with renal abnormalities which often culminate in renal failure. DDD commonly affects children and young adults with a roughly equal male to female preponderance. These patients present with varying degrees of hematuria and/or proteinuria. In addition, serum analysis often but not invariably reveals hypocomplementemia, particularly of the complement protein C3, which has led some researchers to speculate that abnormalities of the complement cascade contribute to the pathogenesis of DDD. To this end, it has been postulated that complement proteins C3b-dependent convertase and factor H play roles in this process by leading to the depletion of C3. 'C3 nephritic factor' and other nephritic factors are autoantibodies which prevent the inactivation of C3b-dependent convertase by factor H. These nephritic factors have been observed in the sera of many (40-70%) patients with DDD, and their presence is often used as a screening tool. In addition, a DDD-like clinical syndrome occurs, interestingly, in Yorkshire pigs which recessively inherit mutations in the factor H gene. These mutations alter factor H and prevent its association with and subsequent inhibition of C3b-dependent convertase. Mutations in the factor H gene are also seen humans; however, those who harbor defective factor H genes do not uniformly develop DDD. Partial lipodystrophy (PL), a disfiguring disorder in which adipose tissue from the upper half of the body is progressively lost, is clinically associated with DDD, the presence of nephritic factors in the serum, and C3 hypocomplementemia. It is known that adipocytes as well as glomerular cells secrete complement proteins. In the presence of nephritic factors, it is proposed that the complement cascade is directly initiated in the vicinity of the cell leading to its lysis. Although such hypotheses serve to unify DDD, C3 hypocomplementemia, the presence of nephritic factors, and PL, other explanations as to the pathophysiology of the renal lesions in DDD are certainly possible.
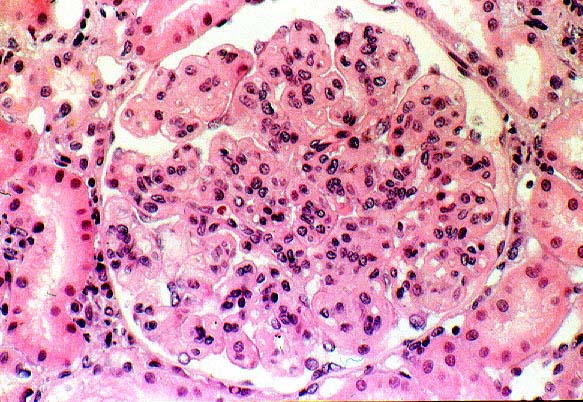
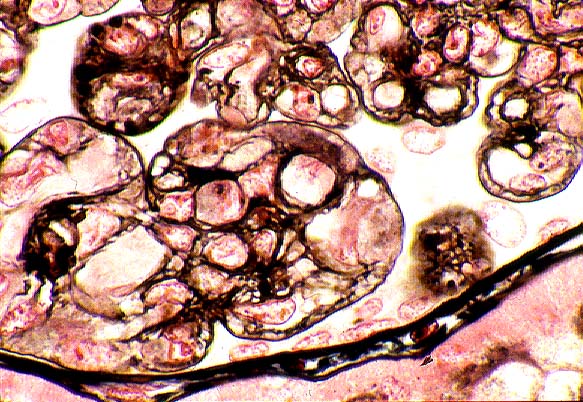
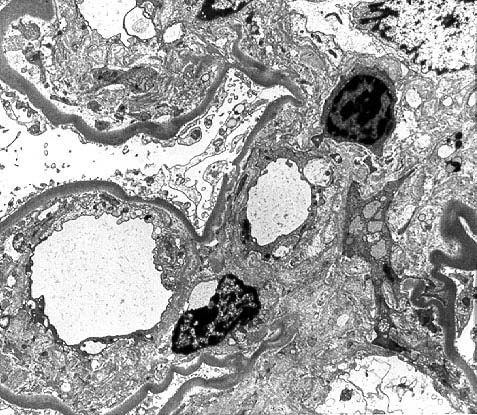
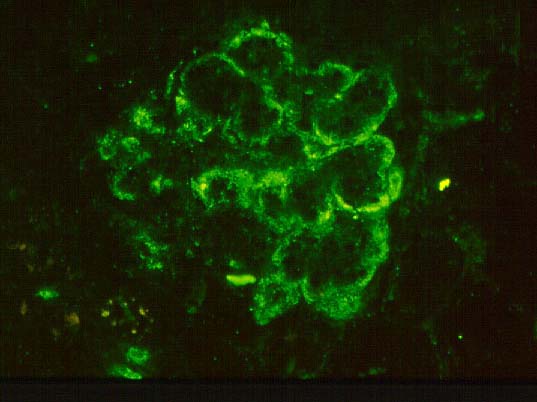
Some of the histologic features of DDD are sufficiently unique to make pathologic diagnosis of DDD relatively straight forward. Histologically, DDD is defined by the presence of irregular electron dense deposits in the lamina densa of the glomerular basement membrane which are observed by electron microscopy. These deposits are strongly PAS positive and are dark blue when stained with toluidine blue. With the fluorescent dye, thioflavine-T, the deposits fluoresce brightly. With methenamine silver stain, the dense deposits appear brown (semi-argyrophilic) while the glomerular basement membrane lining the deposits is intensely argyrophilic. The appearance of the silver-stained glomerular basement membrane bordering the brown deposits is termed 'tram tracking'. Immunofluorescent analysis reveals the presence of the complement protein C3 which produces a characteristic ribbon-like staining pattern of the peripheral capillary loop; however, there is a lack of staining of the dense deposits. Occasionally, segmental staining for IgM with anti-IgM antibodies is seen. Currently, it is unclear what types of molecules comprise the dense deposits. Although the electron microscopic findings are a classic and constant feature of DDD, a wide range of light microscopic patterns may be observed, including membranoproliferative glomerulonephritis (lobular glomerulonephritis), glomerular sclerosis, pure mesangial hypercellularity, membranous glomerulonephritis-like pattern, cresenteric glomerulonephritis, minimal changes, or focal and segmental necrotizing glomerulonephritis. In addition to the glomerular features, abnormalities of the tubules, capillaries and interstitium including the presence of dense deposits may be seen.
Distinguishing DDD from membranoproliferative glomerulonephritis type I (MPGN-I) may be quite difficult clinically since both are characterized by a nephrotic and/or nephritic clinical picture. However, microscopic examination reveals differences that allow their separation. In MPGN-I, most glomeruli demonstrate global hypercellularity in a lobular pattern (IMAGE 10). This differs from DDD in which a variety of light microscopic patterns may be seen, as mentioned above. The capillary walls in the glomeruli are markedly thickened and show intense staining with PAS in MPGN-I. Methenamine silver also stains the glomerular basement membrane of the thickened capillary loops to reveal splitting or tram tracking (IMAGE 11) similar to what is observed in DDD; however, the material deposited between the glomerular basement membrane borders is nonargyrophilic and different from the deposits in DDD. Tram tracking in MPGN-I is caused by an increase in mesangial cells and mesangial matrix in the capillary loops, called 'mesangial interposition', which leads to new subendothelial basement membrane deposition. Electron microscopic findings in MPGN-I show mesangial interposition, subendothelial immune deposits and a new layer of subendothelial basement membrane (IMAGE 12). Immunofluorescence microscopy reveals granular staining for the complement protein C3 (IMAGE 13) which differs from the ribbony pattern seen in DDD. In addition, MPGN-I biopsies, unlike those of DDD, may show staining for complement proteins Clq, C4 and one or more immunoglobulins, particularly IgG and frequently IgM. MPGN-I has a recognized association with chronic hepatitis C infection and cryoglobulinemia in a significant percentage of patients.
Studies have been initiated to predict which clinical and pathological features of DDD are most indicative of a poor outcome (i.e. end-stage renal disease). Clinical findings such as heavy proteinuria, macroscopic hematuria, and sterile pyuria portend a worse outcome for patients. Likewise, certain histopathologic features seen on renal biopsy such as the presence of crescents at initial biopsy, the presence of polymorphonuclear leukocytes in the glomerular capillaries, severe mesangial hypercellularity, and glomerular sclerosis with obliteration of the capillary loop are associated with progressive loss of renal function.
Although diagnosing DDD is fairly easily accomplished through moderm techniques in pathology, preventing its onset or achieving remission once progressive renal disease begins currently alludes physicians. Moreover, the rate of recurrence in renal allografts in patients transplanted for DDD approaches 100%. Thus, additional experimentation is essential to understand its underlying cause and to successfully halt its progression to renal insufficiency in affected individuals.
REFERENCES
![]() Contributed by Marie C. DeFrances, MD, PhD and Sheldon Bastacky, MD
Contributed by Marie C. DeFrances, MD, PhD and Sheldon Bastacky, MD