PATIENT HISTORY:
A 56 year-old right-handed white woman developed left facial numbness. Two days later, she developed right "peripheral" facial weakness. Two weeks later, her left hand became weak without a sensory disturbance. She then developed left-sided hearing loss and tinnitus.
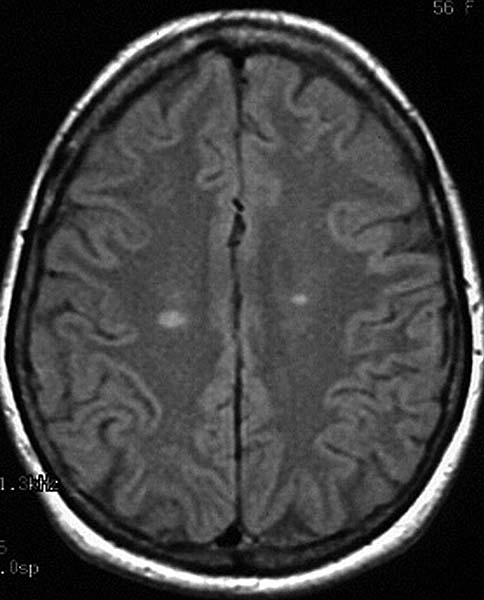
A cranial magnetic resonance (MR) scan (Fig.1) revealed multiple areas of periventricular bright T2 signal with callosal involvement. There was no abnormal enhancement or brainstem abnormality. Lumbar puncture revealed no inflammatory cells and normal protein and glucose. The IgG index was 1.7 (normal < 0.7) with an IgG synthesis rate of 23.8mg per 24h (normal < 3.3). Two oligoclonal bands were present. A diagnosis of probable multiple sclerosis was rendered.
Over the next two weeks, the left facial numbness resolved, but the right facial weakness persisted. Two months after initial onset of symptoms, the patient developed numbness in both feet (especially dorsally, right more than left) and in the perineum as well as difficulty walking. She was noted to have left arm hyperreflexia, decreased pin sensation over the left lateral thigh, and a spastic gait.
Nerve conduction studies of the face and left arm and leg were normal except for a very low amplitude left ulnar motor response, a prolonged median sensory distal latency, and absent H reflexes. In addition, the right tibial motor amplitude was 8mV; the left was 21mV. The left sural amplitude was 50% higher than the right. The right facial motor response was reduced in amplitude and long in latency compared to the left. A needle electromyogram examination was essentially normal. MR imaging of the entire spine was also normal, and a brain MR scan showed no new signal changes. She received 1,000 mg of intravenous methylprednisolone daily for three days and was referred to our care.
She was previously in good health except for irritable bowel syndrome. She had no visual symptoms or Lhermitte's phenomenon. There was no arthritis, rash, or constitutional symptoms. She did not use alcohol and quit smoking shortly after onset of symptoms. Family history was positive for diabetes, hypertension and stroke. Her medications were hyoscyamine sulfate (for irritable bowel symptoms) and aspirin.
Our neurologic examination revealed normal mental status and cranial nerves except for right facial weakness. There was significant weakness of the right triceps and mild weakness in the proximal right lower extremity. There was atrophy and weakness of the abductor digiti minimi and interossei muscles in the left hand. Tendon reflexes were normal and symmetric except for a reduced right triceps jerk. Plantar responses were flexor. Coordination and sensation were normal. She could not hop on the right foot.
Additional laboratory studies revealed that the erythrocyte sedimentation rate, rheumatoid factor, C3, C4, and SS-A, SS-B, anti-nuclear, Lyme, anti-cardiolipin, hepatitis B, and hepatitis C antibodies were normal or negative. Cryoglobulins were present on one occasion, but the results were not reproducible.
The patient was stable until she developed diplopia on upgaze 7 months after onset of the first symptoms. A new right insular lesion was detected by MR scan. Gallium scan and chest computed tomography were normal. The serum angiotensin converting enzyme (ACE) was 15 U/L (normal 9-67). Urine protein electrophoresis revealed trace free lambda Bence Jones protein. A mammogram was normal. Extensive electrodiagnostic studies were then performed and revealed an asymmetric, wide-spread axon loss process consistent with multiple mononeuropathies. Sural nerve and medial gastrocnemius muscle biopsies were performed.