FINAL DIAGNOSIS
Medullary thyroid carcinoma
DISCUSSION
The patient underwent total thyroidectomy with bilateral cervical lymph node dissection and the pathology was signed out as medullary thyroid carcinoma (2.5cm), pT2 N1b M(not applicable). Medullary thyroid carcinoma (MTC) accounts for approximately 5% of thyroid malignancies.(1) In comparison to well-differentiated papillary and follicular thyroid carcinomas, MTCs are clinically more aggressive and thus, accurate diagnosis is extremely important for appropriate management.
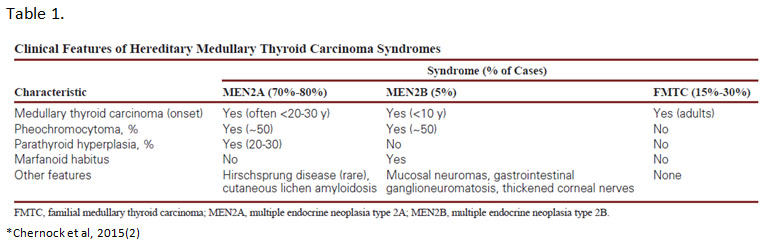
MTC is a neuroendocrine-type carcinoma that is composed of parafollicular C-cells. Classically, MTCs on histology show solid sheets and nests of cells that are plasmacytoid, have abundant pale pink cytoplasm and round to oval, finely-stippled nuclei. However, these tumors can have widely divergent morphologies including spindle cell, papillary, clear cell or oncocytic, among others. In about 20-30% of cases, MTCs are hereditary and are associated with germline RET mutations. These cases occur in the setting of multiple endocrine neoplasia types 2A and 2B (MEN2A/MEN2B) and familial medullary thyroid carcinoma (FMTC) syndromes. Both MEN2A and MEN2B are associated with other endocrine abnormalities whereas FMTC does not have other associated clinical features. (Table 1)
Most cases of MTC (70-80%) are sporadic and about 50% of which will have somatic RET mutations. Of the sporadic MTCs, HRAS and KRAS mutations may also be present and appear to be mutually exclusive with RET mutations. Clinically, MTC typically presents as a single "cold" thyroid nodule. Serum calcitonin and CEA levels are often elevated.
Hereditary Disease
Germline RET mutations are the genetic basis for MEN2A, MEN2B and FMTC.(3) RET encodes for a receptor tyrosine kinase that is important in the development of the parathyroids, urogenital system and neural crest.(4) More than 80 RET mutations in exons 5, 8, 10, 11, 12 to 16 and 19 have been associated with hereditary MTC.(5) Majority are single nucleotide missense mutations that lead to constitutive activation of the RET tyrosine kinase. The specific location of the mutations within the RET gene correlates with the different constellation of phenotypes in each hereditary syndrome. (Figure 2)
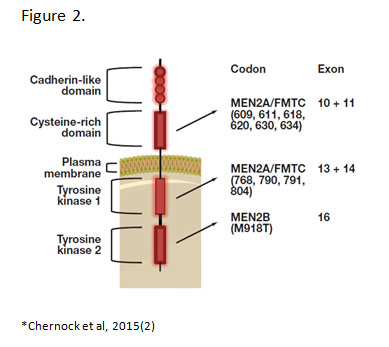
Several RET mutations specifically located in cysteines within the extracellular cysteine-rich domain give rise to MEN2A syndrome.(6) These mutations allow for constitutive dimerization of two RET proteins in the absence of ligand by means of abolishing intramolecular disulfide bridge formation.(7) FMTC mutations are also located in the cysteine-rich domain or TKI domain and overlap with MEN2A mutations which is why FMTC is considered a variant of MEN2A with low penetrance of the other endocrine abnormalities, e.g. pheochromocytoma and hyperparathyroidism. In comparison, MEN2B is caused by a single mutation in exon 16 (M918T) in about 95% of cases. This mutation causes a conformational change in the TK2 binding pocket leading to constitutive kinase activation in the absence of dimerization.(7, 8) The different mechanisms of RET activation trigger different patterns of phosphorylation and cell signaling. This may explain differences in disease phenotypes among the syndromes. Specific RET mutations are also associated with variation in disease phenotypes as well as differing risk levels (MTC aggressiveness) within each of the hereditary syndromes.
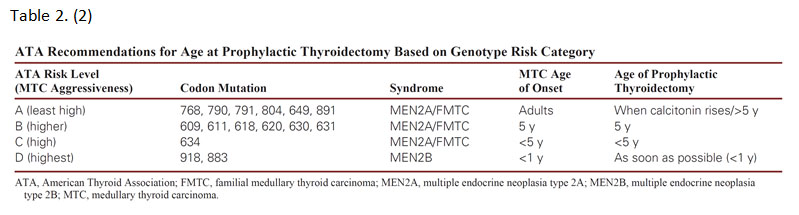
All patients with a personal history of MTC, primary C-cell hyperplasia, or clinical features of MEN2A/MEN2B or FMTC should be offered testing for germline RET mutations.(9) Anyone with a first-degree relative with MEN2 or FMTC should also undergo genetic testing. Prophylactic thyroidectomy may be offered to screened family members harboring a RET mutation before MTC develops. (Table 2) They can then be subsequently monitored for pheochromocytoma or hyperparathyroidism, if indicated by the specific mutation identified.
Sporadic Disease
Sporadic MTCs have somatic RET mutations in about 50% of cases, the most common of which is M918T (75-95%). This mutation (seen in MEN2B) is also associated with more aggressive/advanced stage disease.(10) Activating RAS mutations have also been identified in a subset of sporadic MTCs. They appear to be mutually exclusive with RET mutations and are present in 10-45% of RET wild-type sporadic tumors. Of note, the RAS signaling pathway is activated by the RET tyrosine kinase receptor. Activation of this pathway by either RET or RAS appears to play a major role in both hereditary and sporadic tumors.
Molecular testing has been a mainstay in the clinical management of MTC. Tyrosine kinase inhibitors (TKIs) have been approved for advanced disease and are used in numerous clinical trials. Therefore, genetic testing of tumor tissue is progressively gaining ground and may become part of clinical practice in the near future. For this particular case, the detected allele frequency for the RET mutation was 37% using tumor tissue. This is suggestive of a sporadic mutation (<50% allele frequency). Testing of patient's normal tissue such as peripheral blood can discriminate between germline and acquired mutations.
REFERENCES
![]() Contributed by Remegio J. Maglantay Jr., MD; Yuri E. Nikifirov, MD,PhD; Michael S. Landau, MD
Contributed by Remegio J. Maglantay Jr., MD; Yuri E. Nikifirov, MD,PhD; Michael S. Landau, MD